The Principles of Super-Resolution Fluorescence Microscopy (Review)
Diffraction limit of optical microscopy impedes imaging of biological objects much smaller than the wavelength of light. Conventional fluorescence microscopy does not enable to study fine structure and processes in a living cell at the macromolecular level. Super-resolution fluorescence microscopy techniques that overcome the diffraction barrier have opened up new opportunities for biological and biomedical research. These methods combine the resolution power comparable to electron microscopy with non-invasiveness and labeling specificity of fluorescence imaging. This review describes the modern super-resolution microscopy approaches, their principles and applications. We discuss the key achievements and the main recent trends in this area.
Of all the methods of microscopic analysis fluorescence microscopy is the most widespread due to two main advantages. Firstly, the fluorophores used as contrasting agents can specifically label cell structures. Secondly, fluorescence microscopy is characterized by low invasiveness and allows the observation of living cells in real-time [1–3]. Once fluorescent proteins had been discovered, it became possible to study gene expression, localization and functional activity of proteins in living cells [4, 5].
The combination of fluorescence and confocal systems allows 3D-visualization of cells and tissues [6]. Key discoveries, such as the development of laser scanning confocal microscopy and spinning-disc confocal microscopy, have made the optical microscope one of the most versatile and powerful tools for solving problems in modern cell biology [7, 8].
However, even with high-quality optics, the resolution of a light microscope is limited due to the nature of the light waves. An optical microscope can be regarded as a system of lenses, which provides a magnified image of a small object. When an object is placed in the focal plane, the rays of light from any of its points should converge at a single point on its projection. Light diffraction within the system of microscope lenses leads to a deviation from the laws of geometrical optics [9] and to blurring of the focus spot, preventing the formation of clear images of objects commensurate with the wavelength of the light [10].
To determine the resolution of the optical system various criteria are used, the most well-known being the diffraction limit or Abbe limit. Physically, this implies that the minimum distinguishable size of a point image is limited to the size of the light spot, which is directly proportional to the light wavelength and inversely proportional to twice the numerical aperture of the objective [11, 12].
The theoretical resolution limit under excitation of ultraviolet light (around 400 nm) is about 150 nm in the XY plane and 400 nm on the Z axis, using an oil immersion objective with a numerical aperture of 1.40. Excitation by shorter wavelength light is almost never used because of the photodamage effect of ultraviolet irradiation [13, 14] and the need for more expensive optical components [15]. From a practical point of view, structures that are closer together than 200 nm cannot be optically resolved [10].
Observation of objects smaller than 200 nm, such as viruses, DNA, and mitochondria, has become possible with the development of electron microscopy [16]. A modern electron microscope is characterized by a maximum resolution of up to 0.2 nm, and can provide images of single molecules [17]. However, there are technical features that restrict the widespread use of electron microscopy in biological and biomedical research. While the tedious and multi-step sample preparation procedures can lead to artifacts, affecting the interpretation of images [18, 19], the main drawback of electron microscopy is the lacking opportunity to study processes in living cells [20, 21].
The development of super-resolution light microscopy techniques made it possible to explore living cells at the level of macromolecular complexes [22, 23]. Overcoming the diffraction barrier of traditional light microscope, these methods combine the advantages of electron microscopy and non-invasive fluorescence imaging [20]. In 2014 Eric Betzig, Stefan Hell and William E. Moerner were awarded the Nobel Prize in chemistry for their pioneering work [24, 25].
Near-field microscopy
Imaging of biological sample beyond the diffraction limit was first demonstrated in 1992 using near-field scanning optical microscopy — NSOM (or scanning near-field optical microscopy — SNOM) [26]. Near field is meant the gap close to the surface of the object with the size less than the wavelength of the incident light. NSOM microscopes do not have lenses, the image being obtained by illuminating the object through the small aperture of a submicron optical probe positioned at a distance much shorter than the irradiation wavelength [27]. Super-resolution (up to 20 nm in the lateral plane) [28, 29] is achievable as the near-field illumination is not affected by diffraction or interference effects [30]. Nevertheless, the NSOM method is not widely used in biological studies [31], as it is limited to the visualization of only the surface cell structures, and is technically demanding [32, 33].
The more appropriate method is total internal reflection fluorescence (TIRF) microscopy, based on the effects of evanescent wave in the close proximity to the sample [34]. Excitation of the fluorophores in a region less than 200 nm in thickness provides a high signal-to-noise ratio, while decreasing the photobleaching effect on the fluorophores located in the focal plane. The sum of several sequential TIRF-images obtained using different angles of incidence of the excitation light allows to reconstruct a 3D model of cell structures with axial resolution of up to 20 nm [35]. It is possible to detect signals from individual molecules by TIRF-microscopy [36, 37], however, this method does have its limitations. As NSOM, TIRF-microscopy is effective for the investigation of the cytoskeleton in spread cells, and for processes occurring on the plasma membrane [38, 39], but is not capable for the visualization of intracellular structures [8]. Moreover, TIRF only provides better resolution along the Z axis [40].
Far-field microscopy
The most significant successes in super-resolution imaging have been achieved owing to far-field microscopy, based on the lenses that are relatively remote from the sample [41, 42]. It is worth to mention I5M and 4Pi microscopy, combining two high-aperture lenses for illumination the object from two sides, enabling resolution of up to 100 nm to be obtained along the Z axis [43, 44], even though the lateral resolution is still subject to Abbe’s law.
At the end of the 20th and the beginning of the 21st centuries completely new and revolutionary fluorescence microscopy techniques appeared, able to provide truly sub-diffraction images of biological objects. These techniques can be divided into three major groups: structured illumination microscopy — SIM; stimulated emission depletion microscopy — STED; and single-molecule localization microscopy — SMLM [30]. In these approaches, super-resolution is achieved due to spatial and/or temporal modulation of the fluorophore transition from one molecular state to another, or by reducing the physical size of the point spread function [45].
Structured illumination microscopy (SIM). SIM-microscopy is based on a movable optical grating located in the path of the laser beam [46]. Combining the illumination pattern with the fluorescence pattern of the unknown sample structure, a so-called moiré fringes appear as an interference image with a spatial frequency lower than that of the two initially interacting structures [47]. In the SIM-microscope the striped pattern is shifted or rotated step-by-step before capturing each sequential image. A super-resolution image is then reconstructed by mathematical algorithms [48].
A multicolor 3D-SIM microscope configuration has enabled visualization of eukaryotic cell nucleus demonstrating the structural features of the individual nuclear pore complexes, which previously could be seen only by electron microscopy [49]. 3D-SIM has also provided images of plant cells showing the detailed structure of individual plasmodesmata [50].
The advantages of SIM are its potential to analyze large fields of view, and the compatibility with almost all currently used photostable fluorophores [51]. However, complicated software is required to analyze the various moiré patterns, and there may be artefacts in the super-resolution image. It should be noted that the spatial resolution of the SIM-microscope represents only a two-fold improvement compared to a wide-field microscope, being about 100 and 300 nm in the lateral and axial planes, respectively [10].
Later, saturated structured illumination microscopy (SSIM) has been developed, based on the property of fluorophores for nonlinear increase of emission rate depending on the dose of radiation, besides the moiré effect. This method gives a resolution of up to 50 nm in the XY plane, but it requires a high-power laser, and hence, the use of very photostable fluorophores [52]. Because of the phototoxic effect on cells, SSIM is not suitable for the live-cell imaging [53].
Stimulated emission depletion microscopy (STED). In 1994 a new type of scanning fluorescence microscopy using the effect of stimulated emission depletion (STED) was proposed [54]. This approach is based on reducing the effective diameter of the illuminated spot by an additional STED-laser that suppresses spontaneous emission in the outer rim of the fluorescent spot (Figure 1) due to stimulated emission. The beams of the excitation illumination and the STED-laser are accurately aligned, and the distribution of STED-laser intensities in a focal plane has a “doughnut” shape, with zero intensity in the center. The spontaneous emission of an excited fluorophore (fluorescence) and stimulated emission caused by the STED-laser compete. As a result, at the high power of the STED-laser, only molecules within the zero intensity area fluoresce [55], while in the areas of high intensity there is mainly stimulated emission filtering out due to the coincidence with the STED-laser wavelength. Sequential scanning of the sample provides a complete super-resolution image. In other words, the STED-microscope is a laser scanning confocal microscope equipped with an additional STED-laser, where the sub-diffraction resolution is achieved through selective quenching of the fluorophores [56].
 |
Figure 1. Principles of STED-microscopy. The diagram shows the difference between traditional (top) and STED (bottom) laser scanning confocal microscopy. In traditional microscopy the focus of the excitation laser (blue spot) and the corresponding (green) emission spot are limited by light diffraction, that results in a blurred image (smiley) and the loss of fine details. In STED-microscopy the use of an additional powerful laser irradiating the doughnut-shaped area around the excitation spot (marked with red) causes stimulated emission and, as a result, the reduction of spontaneous emission in this area. Thus, an emission spot, considerably smaller than a diffraction-limited spot, is only observed from the center of the “doughnut” that provides a more detailed image |
The higher the STED-laser power, the better resolution can be obtained. Theoretically, unlimited resolution might be achieved [57], but in practice photodamage of the biological sample limits the power of the STED-beam, so the resolution is usually about 30–80 nm in the XY plane [8].
The use of time-resolved detectors synchronized with the pulse of the excitation laser (gated STED) allows a considerable reduction of the STED-laser power. In these systems, in addition to fluorescence intensity, the lifetime of fluorescent state is taken into account, leading to filtering out of photons whose fluorescence lifetime differs greatly from the expected one. This provides super-resolution and a high signal-to-noise ratio in the image, while lowering the intensity of the STED-laser and hence reducing the risk of photodamage of living objects [58].
STED-based systems appeared to be more popular than near-field microscopy ones, and widely applied to physical [59] and biological issues, especially in the neurobiology field. For examples, STED-microscopy has enabled the visualization of the ultra-thin structures of the dendritic spines of pyramidal neurons in mouse hippocampal slices [60]. The movements of neurotransmitter-containing vesicles in primary cultures of rat neurons has also been described in detail [61]. Subsequently, the successful application ofSTED-microscopyto study the dynamics of neurons in the cerebral cortex of a mouse in vivo was demonstrated [62].
The major advantage of STED-microscopy is that the resolution can be improved in real-time and does not require any long-term data processing.
The main drawbacks of the method are its high cost and the need for high-intensity laser irradiation that limits the number of suitable fluorophores [63]. From a practical point of view, STED-microscopy is effective mainly for fixed samples labeled with photostable fluorescent dyes [64]. Multicolor STED-imaging is extremely difficult as it requires a careful design of the detection channels and the combination of certain dyes [65].
The principle of STED-microscopy can be generalized and represented as a approach using the reversible transition between two states of a molecule — the fluorescent state and the dark one [66]. This concept is manifested in RESOLFT technique (reversible saturable optical linear fluorescence transitions). RESOLFT can be realized through the long-lived dark and fluorescence states of the reversibly photoswitchable fluorophores, using lasers with a special intensity distribution at the focus, as well as in STED microscopy [67]. To initiate such optical transitions within a fluorophore, lower laser power is needed suitable for study of living cells and tissues at a resolution of 50–100 nm [68]. The RESOLFT method has enabled the tracking the dynamic reconstruction of actin in the dendritic spines of living hippocampal neurons over several hours without any signs of tissue destruction, and with a three times greater resolution than that of a confocal microscope [69].
Single-molecule localization microscopy (SMLM). In 2006 three laboratories independently demonstrated a new principle of super-resolution microscopy. The following methods were presented: STORM (stochastic optical reconstruction microscopy) [70], PALM (photoactivated localization microscopy) [71], and FPLAM (fluorescence photoactivation localization microscopy) [72], which, together, laid the foundations for SMLM. The main requirement for SMLM is the detection of single-molecule fluorescence that allows the calculation of the position of an individual molecule with extremely high accuracy (Figure 2 (a)). To obtain data for a multitude of such molecules, fluorophore photoswitching is needed, i.e. the transition between dark and fluorescent states [73]. Unlike STED and RESOLFT, determining the position of fluorescent molecules, on- and off-switching of the molecules is random in SMLM [74, 75]. The microscope detector accumulates the signals from the “on” fluorophore, with the uncertainty of its localization being inversely proportional to the number of detected photons [76].
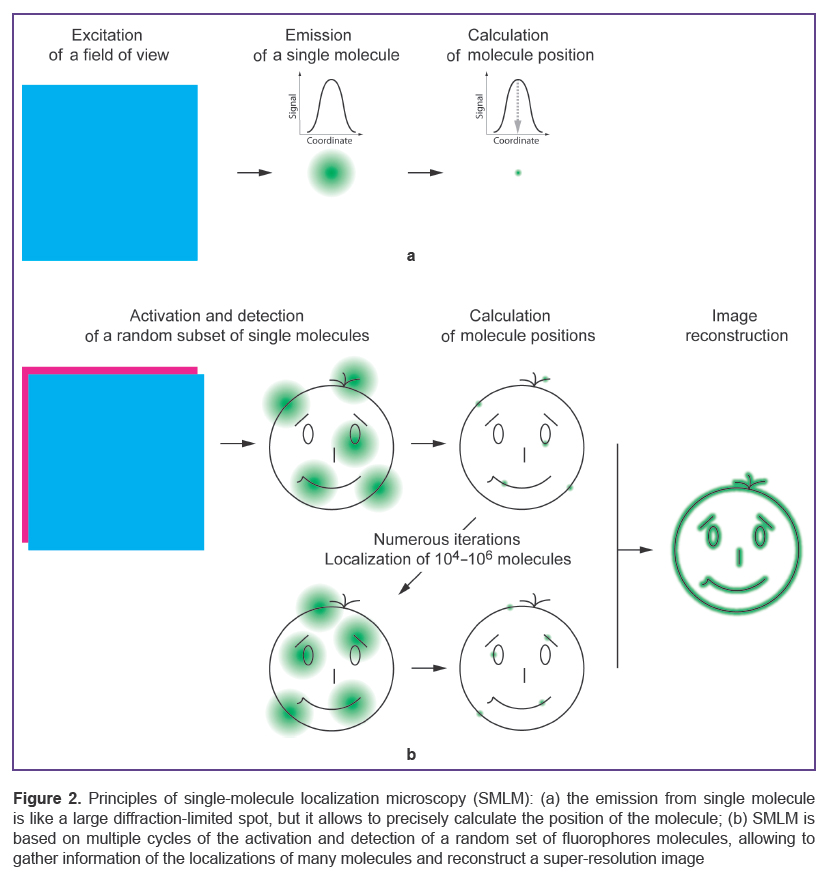
The physical concept of SMLM is the consecutive visualization of subsets of fluorophores scattered across thousands of images in order to map their real distribution density (Figure 2 (b)) [77]. The quality of this reconstruction strongly depends on the algorithm used for data processing. Different software has been developed to recover and localize the centres of point sources of emission [78]. Analysis of the initial data usually involves the identification of individual fluorophores, determination of the coordinates of each fluorophore molecule, and the reconstruction of a super-resolution image, where the positions of all the localized molecules are mapped by overlaying the initial acquired images [79]. The resolving power is commonly defined by multiple measurements of the localization of a single molecule and calculation of the standard deviation from the normal distribution of the data. As an alternative, Fourier ring correlation analysis may be applied to take into account the density and character of the fluorophore distribution by evaluating the similarity of two independent reconstructions from a single initial data source [80, 81].
The localization precision in the lateral plane is usually 20–50 nm, while the resolution on the Z axis varies depending on the microscope configuration [8]. With methods based on two objectives, located on either side of the sample, such as dual-objective-STORM [82] and iPALM (interferometric-PALM) [83], a 20 nm axial resolution power can be achieved; while the standard TIRF-mode only provides a Z axis resolution of 100 nm. To obtain information of the position of a molecule on the Z axis an additional cylindrical lens can be used in front of the detector, so that the image of the point spread function is predictably distorted the greater the distance from the focus [84].
Beyond PALM and STORM, new methods are now being implemented to increase the potential of single-molecule localization microscopy. They include BaLM (bleaching/blinking assisted localization microscopy) [85] and gSHRImP (generalized single-molecule high-resolution imaging with photobleaching) [86], providing resolving powers of 30 and 50 nm, respectively. Their principle is based on the extraction of information of the position of each fluorophore not only from its blinking, but also from its photobleaching, which is random as well. This approach widens the range of suitable fluorescent dyes and allows for generating a pattern of the distribution of single molecules at high density [87].
Besides, SOFI method (super-resolution optical fluctuation imaging) has been proposed, based on the statistical analysis of the temporal fluctuations of fluorophore fluorescence. SOFI-reconstruction is possible through the analysis of fewer images than needed for PALM and STORM, but it requires the absence of any intervals between frames and, as a rule, a high speed of acquisition [88]. The advantages of SOFI are both its ability to analyze high-densely labeled samples and to achieve a considerable reduction of the background signal in the reconstructed image [89].
Among all the super-resolution fluorescence microscopy techniques, SMLM has the best quality/cost ratio. A wide-field microscope equipped with a set of continuous lasers, power of around 100 mW, a high-aperture objective and a sensitive CCD-camera supposed to be enough for implementation. To process the data there is a great variety of free available software such as rapidSTORM [90] or quickPALM [91]. Moreover, some leading manufacturers of optical equipment from Nikon, Zeiss and Leica offer to researchers complete, integrated systems for SMLM [92].
Conclusion
By now, super-resolution microscopy methods remain innovative and have a wide potential in different areas of biology and biomedicine. There is a continuous progress towards development of superior fluorophores, data processing algorithms, and optical setups [93]. For example, an original principle of live-cell 3D imaging has been developed [94], new approaches to labeling protein molecules [95] and data processing [96] have been tested. In recent years, the technologies that combine super-resolution fluorescence microscopy with other methods of visualization have been in active development. Thus, correlated light and electron microscopy — CLEM, becomes very efficient in combination with PALM-based methods allowing the correlation analysis of electron micrographs with fluorescence images at a nanometer scale [97]. The combination of super-resolution and light sheet microscopy (light sheet fluorescence microscopy — LSFM) successfully works as it allows visualization of thick living biological specimens over long periods of time. 3D imaging of living samples with thickness of up to 150 µm at sub-diffraction resolution has been demonstrated by individual molecule localization with selective plane illumination microscopy — IML-SPIM [98]. A technology based on lattice light sheet microscopy — LLSM, aimed at the visualization of rapid dynamic processes within the entire organism, has been developed [99]. The LLSM method has also been successfully combined with PAINT technology, resulting in multicolor 3D images of thick, high-densely labeled samples of dividing cells and embryos of model organisms [100].
A major trend is the development of techniques less damaging living cells and being suitable for the study of dynamic processes. Modified variants of SIM-microscopy with better resolution at high-speed acquisition and lower power of laser irradiation (high-numerical aperture TIRF-SIM and patterned activation nonlinear-SIM) have been demonstrated [101]. There is potential in the complementarity of super-resolution microscopy and microfluidics technology as it allows studying the subcellular processes in living objects within a controlled 3D-microenvironment resembling the physiological conditions [102].
It is important to emphasize that the development of such technologies is only possible through the close collaboration of researchers with different expertises: engineers, physicists, software engineers, mathematicians, biophysicists, chemists, and molecular biologists [103]. On the one hand, the interdisciplinary nature of super-resolution microscopy should facilitate fast progress and breakthrough achievements, but on the other hand, it can restrict its practical implementation. In the near future, super-resolution microscopy techniques are expected to be widely used to solve fundamental and applied issues and become a routine procedure for researchers.
Study Funding. The work was supported by the Russian Science Foundation (project №14-25-00129).
Conflicts of Interest. The authors declare no conflicts of interest.
References
- Periasamy A. Methods in Cellular Imaging. Springer; 2001.
- Ishikawa-Ankerhold H.C., Ankerhold R., Drummen G.P.C. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012; 17(4): 4047–4132, http://dx.doi.org/10.3390/molecules17044047.
- Ettinger A., Wittmann T. Fluorescence live cell imaging. Methods in Cell Biology, vol. 123, 2014, http://dx.doi.org/10.1016/b978-0-12-420138-5.00005-7.
- Lippincott-Schwartz J., Patterson G.H. Development and Use of Fluorescent Protein Markers in Living Cells. Science 2003; 300(5616): 87–91, http://dx.doi.org/10.1126/science.1082520.
- Chudakov D.M., Matz M.V., Lukyanov S., Lukyanov K.A. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol Rev 2010; 90(3): 1103–1163, http://dx.doi.org/10.1152/physrev.00038.2009.
- Lichtman J.W., Conchello J.A. Fluorescence microscopy. Nat Methods 2005; 2(12): 910–919, http://dx.doi.org/10.1038/nmeth817.
- Stehbens S., Pemble H., Murrow L., Wittmann T. Imaging intracellular protein dynamics by spinning disk confocal microscopy. Methods Enzymol 2012; 504: 293–313, http://dx.doi.org/10.1016/B978-0-12-391857-4.00015-X.
- Schermelleh L., Heintzmann R., Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol 2010; 190(2): 165–175, http://dx.doi.org/10.1083/jcb.201002018.
- Gu M. Advanced Optical Imaging Theory. Springer Science & Business Media; 2000.
- Huang B., Bates M., Zhuang X. Super-Resolution Fluorescence Microscopy. Annu Rev Biochem 2009; 78(1): 993–1016, http://dx.doi.org/10.1146/annurev.biochem.77.061906.092014.
- Stelzer E.H.K. Beyond the diffraction limit? Nature 2002; 417(6891): 806–807, http://dx.doi.org/10.1038/417806a.
- Zhang X., Liu Z. Superlenses to overcome the diffraction limit. Nat Mater 2008; 7(6): 435–441, http://dx.doi.org/10.1038/nmat2141.
- Wiedenmann J., Oswald F., Nienhaus G.U. Fluorescent proteins for live cell imaging: Opportunities, limitations, and challenges. IUBMB Life 2009; 61(11): 1029–1042, http://dx.doi.org/10.1002/iub.256.
- Wäldchen S., Lehmann J., Klein T., van de Linde S., Sauer M. Light-induced cell damage in live-cell super-resolution microscopy. Sci Rep 2015; 5: 15348, http://dx.doi.org/10.1038/srep15348.
- Botchway S.W., Scherer K.M., Hook S., Stubbs C.D., Weston E., Bisby R.H., Parker A.W. A series of flexible design adaptations to the Nikon E-C1 and E-C2 confocal microscope systems for UV, multiphoton and FLIM imaging. J Microsc 2015; 258(1): 68–78, http://dx.doi.org/10.1111/jmi.12218.
- Bozzola J.J., Russell L.D. Electron Microscopy: Principles and Techniques for Biologists. Jones & Bartlett Learning; 1999.
- van Heel M., Gowen B., Matadeen R., Orlova E.V., Finn R., Pape T., Cohen D., Stark H., Schmidt R., Schatz M., Patwardhan A. Single-particle electron cryo-microscopy: towards atomic resolution. Q Rev Biophys 2000; 33(4): 307–369.
- Ayache J., Beaunier L., Boumendil J., Ehret G., Laub D. Sample Preparation Handbook for Transmission Electron Microscopy: Methodology. Springer Science & Business Media; 2010.
- Hayat M. Fixation for Electron Microscopy. Elsevier; 2012.
- Henriques R., Griffiths C., Hesper Rego E., Mhlanga M.M. PALM and STORM: unlocking live-cell super-resolution. Biopolymers 2011; 95(5): 322–331, http://dx.doi.org/10.1002/bip.21586.
- Leung B.O., Chou K.C. Review of Super-Resolution Fluorescence Microscopy for Biology. Appl Spectrosc 2011; 65(9): 967–980, http://dx.doi.org/10.1366/11-06398.
- Klein T., Proppert S., Sauer M. Eight years of single-molecule localization microscopy. Histochem Cell Biol 2014; 141(6): 561–575, http://dx.doi.org/10.1007/s00418-014-1184-3.
- Sydor A.M., Czymmek K.J., Puchner E.M., Mennella V. Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol 2015; 25(12): 730–748, http://dx.doi.org/10.1016/j.tcb.2015.10.004.
- Möckl L., Lamb D.C., Bräuchle C. Super-resolved Fluorescence Microscopy: Nobel Prize in Chemistry 2014 for Eric Betzig, Stefan Hell, and William E. Moerner. Angew Chem Int Ed 2014; 53(51): 13972–13977, http://dx.doi.org/10.1002/anie.201410265.
- Stelzer E.H.K. Better Imaging through Chemistry. Cell 2014; 159(6): 1243–1246, http://dx.doi.org/10.1016/j.cell.2014.11.032.
- Betzig E., Trautman J.K. Near-field optics: microscopy, spectroscopy, and surface modification beyond the diffraction limit. Science 1992; 257(5067): 189–195, http://dx.doi.org/10.1126/science.257.5067.189.
- Dunn R.C. Near-field scanning optical microscopy. Chem Rev 1999; 99(10): 2891–2928, http://dx.doi.org/10.1021/cr980130e.
- Alù A., Engheta N. Cloaked Near-Field Scanning Optical Microscope Tip for Noninvasive Near-Field Imaging. Phys Rev Lett 2010; 105(26), http://dx.doi.org/10.1103/physrevlett.105.263906.
- Oshikane Y., Kataoka T., Okuda M., Hara S., Inoue H., Nakano M. Observation of nanostructure by scanning near-field optical microscope with small sphere probe. Sci Technol Adv Mater 2007; 8(3): 181–185, http://dx.doi.org/10.1016/j.stam.2007.02.013.
- Fernández-Suárez M., Ting A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol 2008; 9(12): 929–943, http://dx.doi.org/10.1038/nrm2531.
- Han R., Li Z., Fan Y., Jiang Y. Recent Advances in Super-Resolution Fluorescence Imaging and Its Applications in Biology. J Genet Genomics 2013; 40(12): 583–595, http://dx.doi.org/10.1016/j.jgg.2013.11.003.
- Dickenson N.E., Armendariz K.P., Huckabay H.A., Livanec P.W., Dunn R.C. Near-field scanning optical microscopy: a tool for nanometric exploration of biological membranes. Anal Bioanal Chem 2010; 396(1): 31–43, http://dx.doi.org/10.1007/s00216-009-3040-1.
- van Zanten T.S., Cambi A., Garcia-Parajo M.F. A nanometer scale optical view on the compartmentalization of cell membranes. Biochim Biophys Acta 2010; 1798(4): 777–787, http://dx.doi.org/10.1016/j.bbamem.2009.09.012.
- Yamamura H., Suzuki Y., Imaizumi Y. New light on ion channel imaging by total internal reflection fluorescence (TIRF) microscopy. J Pharmacol Sci 2015; 128(1): 1–7, http://dx.doi.org/10.1016/j.jphs.2015.04.004.
- Fu Y., Winter P.W., Rojas R., Wang V., McAuliffe M., Patterson G.H. Axial superresolution via multiangle TIRF microscopy with sequential imaging and photobleaching. Proc Natl Acad Sci U S A 2016; 113(16): 4368–4373, http://dx.doi.org/10.1073/pnas.1516715113.
- Axelrod D. Total Internal Reflection Fluorescence Microscopy in Cell Biology. Traffic 2001; 2(11): 764–774, http://dx.doi.org/10.1034/j.1600-0854.2001.21104.x.
- Kudalkar E.M., Davis T.N., Asbury C.L. Single-Molecule Total Internal Reflection Fluorescence Microscopy. Cold Spring Harb Protoc 2016; 2016(5): db.top077800, http://dx.doi.org/10.1101/pdb.top077800.
- Jaiswal J.K., Simon S.M. Imaging single events at the cell membrane. Nat Chem Biol 2007; 3(2): 92–98, http://dx.doi.org/10.1038/nchembio855.
- Rappoport J.Z. Focusing on clathrin-mediated endocytosis. Biochem J 2008; 412(3): 415–423, http://dx.doi.org/10.1042/BJ20080474.
- Chung E., Kim D., Cui Y., Kim Y.H., So P.T.C. Two-Dimensional Standing Wave Total Internal Reflection Fluorescence Microscopy: Superresolution Imaging of Single Molecular and Biological Specimens. Biophys J 2007; 93(5): 1747–1757, http://dx.doi.org/10.1529/biophysj.106.097907.
- Eggeling C., Willig K.I., Sahl S.J., Hell S.W. Lens-based fluorescence nanoscopy. Q Rev Biophys 2015; 48(2): 178–243, http://dx.doi.org/10.1017/s0033583514000146.
- Lippincott-Schwartz J., Manley S. Putting super-resolution fluorescence microscopy to work. Nat Methods 2009; 6(1): 21–23, http://dx.doi.org/10.1038/nmeth.f.233.
- Dyba M., Marcus D., Hell S.W. Focal Spots of Size λ / 23 Open Up Far-Field Florescence Microscopy at 33 nm Axial Resolution. Phys Rev Lett 2002; 88(16), http://dx.doi.org/10.1103/physrevlett.88.163901.
- Bewersdorf J., Schmidt R., Hell S.W. Comparison of I5M and 4Pi-microscopy. J Microsc 2006; 222(Pt 2): 105–117, http://dx.doi.org/10.1111/j.1365-2818.2006.01578.x.
- Nienhaus K., Nienhaus G.U. Where Do We Stand with Super-Resolution Optical Microscopy? J Mol Biol 2016; 428(2 Pt A): 308–322, http://dx.doi.org/10.1016/j.jmb.2015.12.020.
- Gustafsson M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 2000; 198(Pt 2): 82–87.
- Yamanaka M., Smith N.I., Fujita K. Introduction to super-resolution microscopy. Microscopy 2014; 63(3): 177–192, http://dx.doi.org/10.1093/jmicro/dfu007.
- Komis G., Mistrik M., Šamajová O., Ovečka M., Bartek J., Šamaj J. Superresolution live imaging of plant cells using structured illumination microscopy. Nat Protoc 2015; 10(8): 1248–1263, http://dx.doi.org/10.1038/nprot.2015.083.
- Schermelleh L., Carlton P.M., Haase S., Shao L., Winoto L., Kner P., Burke B., Cardoso M.C., Agard D.A., Gustafsson M.G.L., Leonhardt H., Sedat J.W. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008; 320(5881): 1332–1336, http://dx.doi.org/10.1126/science.1156947.
- Fitzgibbon J., Bell K., King E., Oparka K. Super-resolution imaging of plasmodesmata using three-dimensional structured illumination microscopy. Plant Physiol 2010; 153(4): 1453–1463, http://dx.doi.org/10.1104/pp.110.157941.
- Lakadamyali M. Super-resolution microscopy: going live and going fast. Chemphyschem 2014; 15(4): 630–636, http://dx.doi.org/10.1002/cphc.201300720.
- Gustafsson M.G.L. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A 2005; 102(37): 13081–13086, http://dx.doi.org/10.1073/pnas.0406877102.
- Zhang H., Zhao M., Peng L. Nonlinear structured illumination microscopy by surface plasmon enhanced stimulated emission depletion. Opt Express 2011; 19(24): 24783–24794, http://dx.doi.org/10.1364/OE.19.024783.
- Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 1994; 19(11): 780–782.
- Blom H., Widengren J. STED microscopy — towards broadened use and scope of applications. Curr Opin Chem Biol 2014; 20: 127–133, http://dx.doi.org/10.1016/j.cbpa.2014.06.004.
- Willig K.I., Keller J., Bossi M., Hell S.W. STED microscopy resolves nanoparticle assemblies. New J Phys 2006; 8(6): 106–106, http://dx.doi.org/10.1088/1367-2630/8/6/106.
- Harke B., Keller J., Ullal C.K., Westphal V., Schönle A., Hell S.W. Resolution scaling in STED microscopy. Opt Express 2008; 16(6): 4154–4162, http://dx.doi.org/10.1364/OE.16.004154.
- Vicidomini G., Moneron G., Han K.Y., Westphal V., Ta H., Reuss M., Engelhardt J., Eggeling C., Hell S.W. Sharper low-power STED nanoscopy by time gating. Nat Methods 2011; 8(7): 571–573, http://dx.doi.org/10.1038/nmeth.1624.
- Rittweger E., Han K.Y., Irvine S.E., Eggeling C., Hell S.W. STED microscopy reveals crystal colour centres with nanometric resolution. Nat Photonics 2009; 3(3): 144–147, http://dx.doi.org/10.1038/nphoton.2009.2.
- Nägerl U.V., Willig K.I., Hein B., Hell S.W., Bonhoeffer T. Live-cell imaging of dendritic spines by STED microscopy. Proc Natl Acad Sci U S A 2008; 105(48): 18982–18987, http://dx.doi.org/10.1073/pnas.0810028105.
- Lauterbach M.A., Keller J., Schönle A., Kamin D., Westphal V., Rizzoli S.O., Hell S.W. Comparing video-rate STED nanoscopy and confocal microscopy of living neurons. J Biophotonics 2010; 3(7): 417–424, http://dx.doi.org/10.1002/jbio.201000038.
- Berning S., Willig K.I., Steffens H., Dibaj P., Hell S.W. Nanoscopy in a Living Mouse Brain. Science 2012; 335(6068): 551–551, http://dx.doi.org/10.1126/science.1215369.
- Neupane B., Ligler F.S., Wang G. Review of recent developments in stimulated emission depletion microscopy: applications on cell imaging. J Biomed Opt 2014; 19(8): 080901, http://dx.doi.org/10.1117/1.JBO.19.8.080901.
- Combs C.A. Fluorescence microscopy: a concise guide to current imaging methods. Curr Protoc Neurosci 2010; Chapter 2: Unit2.1, http://dx.doi.org/10.1002/0471142301.ns0201s50.
- Bückers J., Wildanger D., Vicidomini G., Kastrup L., Hell S.W. Simultaneous multi-lifetime multi-color STED imaging for colocalization analyses. Opt Express 2011; 19(4): 3130–3143, http://dx.doi.org/10.1364/OE.19.003130.
- Hell S.W. Toward fluorescence nanoscopy. Nat Biotechnol 2003; 21(11): 1347–1355, http://dx.doi.org/10.1038/nbt895.
- Schwentker M.A., Bock H., Hofmann M., Jakobs S., Bewersdorf J., Eggeling C., Hell S.W. Wide-field subdiffraction RESOLFT microscopy using fluorescent protein photoswitching. Microsc Res Tech 2007; 70(3): 269–280, http://dx.doi.org/10.1002/jemt.20443.
- Hofmann M., Eggeling C., Jakobs S., Hell S.W. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci U S A 2005; 102(49): 17565–17569, http://dx.doi.org/10.1073/pnas.0506010102.
- Testa I., Urban N.T., Jakobs S., Eggeling C., Willig K.I., Hell S.W. Nanoscopy of Living Brain Slices with Low Light Levels. Neuron 2012; 75(6): 992–1000, http://dx.doi.org/10.1016/j.neuron.2012.07.028.
- Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 2006; 3(10): 793–796, http://dx.doi.org/10.1038/nmeth929.
- Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S., Davidson M.W., Lippincott-Schwartz J., Hess H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006; 313(5793): 1642–1645, http://dx.doi.org/10.1126/science.1127344.
- Hess S.T., Girirajan T.P.K., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 2006; 91(11): 4258–4272, http://dx.doi.org/10.1529/biophysj.106.091116.
- Sengupta P., Van Engelenburg S., Lippincott-Schwartz J. Visualizing Cell Structure and Function with Point-Localization Superresolution Imaging. Dev Cell 2012; 23(6): 1092–1102, http://dx.doi.org/10.1 016/j.devcel.2012.09.022.
- Vogelsang J., Steinhauer C., Forthmann C., Stein I.H., Person-Skegro B., Cordes T., Tinnefeld P. Make them blink: probes for super-resolution microscopy. Chemphyschem 2010; 11(12): 2475–2490, http://dx.doi.org/10.1002/cphc.201000189.
- Tam J., Merino D. Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J Neurochem 2015; 135(4): 643–658, http://dx.doi.org/10.1111/jnc.13257.
- Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys J 2002; 82(5): 2775–2783, http://dx.doi.org/10.1016/S0006-3495(02)75618-X.
- Almada P., Culley S., Henriques R. PALM and STORM: Into large fields and high-throughput microscopy with sCMOS detectors. Methods 2015; 88: 109–121, http://dx.doi.org/10.1016/j.ymeth.2015.06.004.
- Sage D., Kirshner H., Pengo T., Stuurman N., Min J., Manley S., Unser M. Quantitative evaluation of software packages for single-molecule localization microscopy. Nat Methods 2015; 12(8): 717–724, http://dx.doi.org/10.1038/nmeth.3442.
- Li Y., Ishitsuka Y., Hedde P.N., Nienhaus G.U. Fast and efficient molecule detection in localization-based super-resolution microscopy by parallel adaptive histogram equalization. ACS Nano 2013; 7(6): 5207–5214, http://dx.doi.org/10.1021/nn4009388.
- Endesfelder U., Heilemann M. Art and artifacts in single-molecule localization microscopy: beyond attractive images. Nat Methods 2014; 11(3): 235–238, http://dx.doi.org/10.1038/nmeth.2852.
- Banterle N., Bui K.H., Lemke E.A., Beck M. Fourier ring correlation as a resolution criterion for super-resolution microscopy. J Struct Biol 2013; 183(3): 363–367, http://dx.doi.org/10.1016/j.jsb.2013.05.004.
- Xu K., Babcock H.P., Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods 2012; 9(2): 185–188, http://dx.doi.org/10.1038/nmeth.1841.
- Shtengel G., Galbraith J.A., Galbraith C.G., Lippincott-Schwartz J., Gillette J.M., Manley S., Sougrat R., Waterman C.M., Kanchanawong P., Davidson M.W., Fetter R.D., Hess H.F. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci U S A 2009; 106(9): 3125–3130, http://dx.doi.org/10.1073/pnas.0813131106.
- Huang B., Wang W., Bates M., Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008; 319(5864): 810–813, http://dx.doi.org/10.1126/science.1153529.
- Burnette D.T., Sengupta P., Dai Y., Lippincott-Schwartz J., Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci U S A 2011; 108(52): 21081–21086, http://dx.doi.org/10.1073/pnas.1117430109.
- Simonson P.D., Rothenberg E., Selvin P.R. Correction to Single-Molecule-Based Super-Resolution Images in the Presence of Multiple Fluorophores. Nano Lett 2013; 13(3): 1366–1366, http://dx.doi.org/10.1021/nl4004267.
- Wang Y., Kuang C., Cai H., Li S., Liu W., Hao X., Ge J., Liu X. Sub-diffraction imaging with confocal fluorescence microscopy by stochastic photobleaching. Opt Commun 2014; 312: 62–67, http://dx.doi.org/10.1016/j.optcom.2013.09.022.
- Dertinger T., Colyer R., Iyer G., Weiss S., Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc Natl Acad Sci U S A 2009; 106(52): 22287–22292, http://dx.doi.org/10.1073/pnas.0907866106.
- Geissbuehler S., Dellagiacoma C., Lasser T. Comparison between SOFI and STORM. Biomed Opt Express 2011; 2(3): 408–420, http://dx.doi.org/10.1364/BOE.2.000408.
- Wolter S., Löschberger A., Holm T., Aufmkolk S., Dabauvalle M.C., van de Linde S., Sauer M. rapidSTORM: accurate, fast open-source software for localization microscopy. Nat Methods 2012; 9(11): 1040–1041, http://dx.doi.org/10.1038/nmeth.2224.
- Henriques R., Lelek M., Fornasiero E.F., Valtorta F., Zimmer C., Mhlanga M.M. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods 2010; 7(5): 339–340, http://dx.doi.org/10.1038/nmeth0510-339.
- Sauer M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci 2013; 126(Pt 16): 3505–3513, http://dx.doi.org/10.1242/jcs.123612.
- Cox S., Jones G.E. Imaging cells at the nanoscale. Int J Biochem Cell Biol 2013; 45(8): 1669–1678, http://dx.doi.org/10.1016/j.biocel.2013.05.010.
- Geissbuehler S., Sharipov A., Godinat A., Bocchio N.L., Sandoz P.A., Huss A., Jensen N.A., Jakobs S., Enderlein J., Gisou van der Goot F., Dubikovskaya E.A., Lasser T., Leutenegger M. Live-cell multiplane three-dimensional super-resolution optical fluctuation imaging. Nat Commun 2014; 5: 5830, http://dx.doi.org/10.1038/ncomms6830.
- Ries J., Kaplan C., Platonova E., Eghlidi H., Ewers H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods 2012; 9(6): 582–584, http://dx.doi.org/10.1038/nmeth.1991.
- Levet F., Hosy E., Kechkar A., Butler C., Beghin A., Choquet D., Sibarita J.B. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods 2015; 12(11): 1065–1071, http://dx.doi.org/10.1038/nmeth.3579.
- de Souza N. Super-resolution CLEM. Nat Methods 2014; 12(1): 37–37, http://dx.doi.org/10.1038/nmeth.3239.
- Zanacchi F.C., Lavagnino Z., Donnorso M.P., Del Bue A., Furia L., Faretta M., Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat Methods 2011; 8(12): 1047–1049, http://dx.doi.org/10.1038/nmeth.1744.
- Chen B.C., Legant W.R., Wang K., Shao L., Milkie D.E., Davidson M.W., Janetopoulos C., Wu X.S., Hammer J.A. 3rd, Liu Z., English B.P., Mimori-Kiyosue Y., Romero D.P., Ritter A.T., Lippincott-Schwartz J., Fritz-Laylin L., Mullins R.D., Mitchell D.M., Bembenek J.N., Reymann A.C., Böhme R., Grill S.W., Wang J.T., Seydoux G., Tulu U.S., Kiehart D.P., Betzig E. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 2014; 346(6208): 1257998, http://dx.doi.org/10.1126/science.1257998.
- Legant W.R., Shao L., Grimm J.B., Brown T.A., Milkie D.E., Avants B.B., Lavis L.D., Betzig E. High-density three-dimensional localization microscopy across large volumes. Nat Methods 2016; 13(4): 359–365, http://dx.doi.org/10.1038/nmeth.3797.
- Li D., Shao L., Chen B.C., Zhang X., Zhang M., Moses B., Milkie D.E., Beach J.R., Hammer J.A. 3rd, Pasham M., Kirchhausen T., Baird M.A., Davidson M.W., Xu P., Betzig E. ADVANCED IMAGING. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 2015; 349(6251): aab3500, http://dx.doi.org/10.1126/science.aab3500.
- Chin L.K., Lee C.H., Chen B.C. Imaging live cells at high spatiotemporal resolution for lab-on-a-chip applications. Lab Chip 2016, http://dx.doi.org/10.1039/c5lc01556a.
- Whelan D.R., Bell T.D.M. Super-Resolution Single-Molecule Localization Microscopy: Tricks of the Trade. J Phys Chem Lett 2015; 6(3): 374–382, http://dx.doi.org/10.1021/jz5019702.









