Принципы флюоресцентной микроскопии cверхвысокого разрешения (обзор)
Дифракционный предел разрешения светового микроскопа препятствует наблюдению биологических объектов, размеры которых меньше длины световой волны. Традиционная флюоресцентная микроскопия не позволяет исследовать ультраструктуру, а также процессы, протекающие в живой клетке на уровне макромолекулярных комплексов. Разработка методов высокоразрешающей флюоресцентной микроскопии, преодолевающих дифракционный предел, открыла новые возможности для исследований в сфере биологии и биомедицины. Эти технологии совмещают в себе разрешение, сопоставимое с электронной микроскопией, с неинвазивностью и специфическим мечением, присущими флюоресцентному имиджингу. Рассмотрены современные методы флюоресцентной микроскопии сверхвысокого разрешения, описаны их принципы и области применения. Отмечены ключевые достижения и тенденции развития технологий высокоразрешающей флюоресцентной микроскопии.
Среди разновидностей методов микроскопического анализа наибольшее распространение получила флюоресцентная микроскопия благодаря двум основным преимуществам. Во-первых, в качестве контрастирующих агентов используются флюорофоры, которые обеспечивают специфическое мечение клеточных структур. Во-вторых, флюоресцентная микроскопия характеризуется низкой инвазивностью и позволяет наблюдать за живой клеткой в режиме реального времени [1–3]. С открытием флюоресцентных белков стало доступным изучение экспрессии генов, локализации и функциональной активности белков в живой клетке [4, 5].
Комбинация флюоресценции и конфокальных систем позволила проводить 3D-визуализацию клеток и тканей [6]. Такие ключевые достижения, как разработка лазерной сканирующей конфокальной микроскопии и спиннинг-диск конфокальной микроскопии, сделали оптический микроскоп одним из мощнейших и универсальных инструментов для решения задач современной клеточной биологии [7, 8].
Однако даже при использовании высококачественной оптики разрешающая способность светового микроскопа имеет ограничение, обусловленное самой природой световой волны. Оптический микроскоп можно рассматривать как систему линз, дающих увеличенное изображение малого объекта. При размещении объекта в фокальной плоскости световые лучи от каждой его точки должны также сходиться в единую точку на его проекции. Дифракция света в системе линз микроскопа приводит к отклонению от законов геометрической оптики [9] и размытию сфокусированного пятна, что не позволяет получить отчетливые изображения объектов, соизмеримых с длиной световой волны [10].
Для определения разрешающей способности оптических систем применяются различные критерии, из которых наиболее известен дифракционный предел, или предел Аббе. Его физический смысл заключается в том, что минимальный различимый размер точечного изображения ограничен размером светового пятна, который прямо пропорционален длине волны света и обратно пропорционален двукратной величине числовой апертуры объектива [11, 12].
Теоретический предел разрешения на длине волны возбуждающего ультрафиолетового света (около 400 нм) составляет примерно 150 нм в плоскости xy и 400 нм — по оси Z при использовании объектива с масляной иммерсией и числовой апертурой, равной 1,40. Более короткие длины волн возбуждения практически не используются в силу фотоповреждающего действия ультрафиолетового излучения [13, 14], а также из-за необходимости применения более дорогих оптических компонентов [15]. С практической точки зрения структуры, находящиеся ближе чем 200 нм друг от друга, не могут быть оптически разрешены [10].
Наблюдение объектов размером меньше 200 нм, таких как вирусы, ДНК, митохондрии, стало возможным с появлением электронной микроскопии [16]. Современный электронный микроскоп характеризуется максимальной разрешающей способностью — до 0,2 нм, что позволяет получать изображения отдельных молекул [17]. Однако данный метод имеет ряд технических особенностей, которые затрудняют его широкое распространение в биологических и биомедицинских исследованиях. Трудоемкая и многостадийная процедура приготовления образцов может приводить к появлению артефактов при интерпретации изображений [18, 19]. Основной же недостаток электронной микроскопии заключается в отсутствии возможности изучения процессов, протекающих в живой клетке [20, 21].
Возможность исследовать живую клетку на уровне макромолекулярных комплексов появилась благодаря созданию и развитию технологий высокоразрешающей световой микроскопии [22, 23]. Данные методы, преодолевая дифракционный предел разрешения традиционного светового микроскопа, объединяют в себе преимущества электронной микроскопии и неинвазивного флюоресцентного имиджинга [20]. В 2014 г. за пионерские работы в данной области Эрик Бетциг, Штефан Хелл и Уильям Мёрнер были удостоены Нобелевской премии по химии [24, 25].
Ближнепольная микроскопия
Преодоление дифракционного предела на примере исследования биологического образца впервые было продемонстрировано в 1992 г. с помощью метода сканирующей микроскопии ближнего поля (near-field scanning optical microscopy — NSOM, или scanning near-field optical microscopy — SNOM) [26]. Под ближним полем понимается зона над поверхностью изучаемого объекта размером меньше длины волны падающего света. В микроскопах по типу NSOM нет линз, а изображение получается с помощью освещения объекта через малую диафрагму субмикронного оптического зонда, расположенного на расстоянии много меньше длины волны излучения [27]. Сверхвысокое разрешение (до 20 нм в латеральной плоскости) [28, 29] достигается за счет того, что в области ближнего поля распространение света не подвержено дифракционным или интерференционным эффектам [30]. Тем не менее метод NSOM не нашел широкого применения в биологических исследованиях [31], так как ограничен визуализацией поверхностных клеточных структур и технически сложен [32, 33].
Более пригодным методом стала флюоресцентная микроскопия полного внутреннего отражения (total internal reflection fluorescence — TIRF), основанная на использовании свойств индуцированной быстрозатухающей волны, которая возникает в зоне, близко прилегающей к исследуемому образцу [34]. Возбуждение флюорофоров происходит в области толщиной не более 200 нм, при этом значительно повышается отношение сигнал-шум, а эффект фотообесцвечивания флюорофоров, находящихся вне фокальной плоскости, напротив, снижается. Показано, что комбинация нескольких последовательных TIRF-изображений, полученных при разном угле падения возбуждающего света, позволяет реконструировать 3D-модель клеточных структур с аксиальным разрешением до 20 нм [35]. В условиях TIRF-микроскопии возможно детектирование сигнала от одиночных молекул [36, 37], однако этот метод имеет свои ограничения. Так же, как и NSOM, TIRF-микроскопия эффективна для исследования цитоскелета распластанных клеток и процессов, протекающих на плазматической мембране [38, 39], но неприменима для визуализации внутриклеточных структур [8]. Кроме того, TIRF дает улучшение разрешения только вдоль оси Z [40].
Микроскопия дальнего поля
Наиболее значительные успехи в визуализации со сверхвысоким разрешением были достигнуты благодаря микроскопии дальнего поля, в которой используются линзы, относительно удаленные от образца [41, 42]. Стоит упомянуть о микроскопии I5M и 4Pi, в которой применяют два высокоапертурных объектива для освещения объекта с двух сторон и тем самым добиваются разрешения до 100 нм по оси Z [43, 44]. Тем не менее латеральное разрешение по-прежнему остается подчинено закону Аббе.
В конце XX — начале XXI в. появились принципиально новые, революционные технологии флюоресцентной микроскопии, дающие действительно субдифракционные изображения биологического объекта. Среди них можно выделить три большие группы: микроскопия структурированного освещения (structured illumination microscopy — SIM), микроскопия на основе истощения (флюоресценции) вынужденным излучением (stimulated emission depletion — STED) и локализационная микроскопия одиночных молекул (single-molecule localization microscopy, SMLM) [30]. В этих технологиях сверхвысокое разрешение достигается либо за счет пространственного и/или временного модулирования перехода флюорофора из одного молекулярного состояния в другое, либо за счет уменьшения физического размера функции рассеяния точки [45].
Метод микроскопии структурированного освещения (SIM). SIM-микроскопия базируется на использовании подвижных оптических решеток, размещенных на пути лазерного пучка [46]. При совмещении известного паттерна освещения с неизвестной структурой исследуемого флюоресцентного образца возникает так называемый муаровый узор — интерференционная картина с пространственной частотой ниже, чем у двух изначально взаимодействующих структур [47]. В SIM-микроскопе решетчатая структура смещается или поворачивается поэтапно перед захватом каждого последующего изображения. Затем посредством математических алгоритмов реконструируется высокоразрешенное изображение [48].
С помощью конфигурации многоцветной 3D-SIM-микроскопии была визуализирована структура ядра эукариотической клетки с выявлением особенностей строения единичных комплексов ядерной поры, которые прежде можно было наблюдать только в электронном микроскопе [49]. Также с помощью 3D-SIM были получены изображения растительной клетки, демонстрирующие детальную структуру индивидуальных плазмодесм [50].
Преимущество метода SIM заключается в возможности быстро анализировать большие поля зрения; кроме того, для него пригодны практически все современные флюоресцентные метки, устойчивые к фотообесцвечиванию [51]. Однако для анализа множества муаровых изображений требуются сложные компьютерные программы и на высокоразрешенном изображении могут наблюдаться артефакты. Стоит учитывать, что пространственное разрешение в SIM-микроскопе может быть улучшено лишь в 2 раза по сравнению с широкопольным микроскопом и составляет примерно 100 и 300 нм в латеральной и аксиальной плоскостях соответственно [10].
Позднее была разработана технология микроскопии насыщающего структурированного освещения (saturated structured illumination microscopy — SSIM), основанная помимо муарового эффекта на способности флюорофоров к нелинейному возрастанию эмиссии в зависимости от дозы облучения. Этот метод дает разрешение до 50 нм в плоскости xy, однако требует высокой мощности лазера и, как следствие, флюорофоров с высокой фотостабильностью [52]. В результате вследствие фототоксического воздействия на клетки SSIM не пригодна для имиджинга живых образцов [53].
Микроскопия на основе истощения (флюоресценции) вынужденным излучением (STED). В 1994 г. был предложен новый тип сканирующей флюоресцентной микроскопии STED на основе эффекта подавления спонтанного испускания [54]. Данный подход основан на уменьшении диаметра светящейся точки при помощи дополнительного STED-лазера, который подавляет спонтанную эмиссию во внешней области флюоресцентного пятна (рис. 1) за счет эффекта вынужденного излучения. Пучки возбуждающего и STED-лазера тщательно выравниваются, причем распределение интенсивности STED-лазера в фокусе имеет форму «пончика», с нулевой интенсивностью в центре. Спонтанное излучение возбужденного флюорофора (флюоресценция) и вынужденное излучение за счет STED-лазера конкурируют между собой. В результате при больших интенсивностях STED-лазера флюоресцируют только те молекулы, которые расположены близко к области с нулевой интенсивностью [55], в то время как в зоне высокой интенсивности происходит преимущественно вынужденное излучение флюорофоров, отбрасываемое в оптическом пути из-за совпадения с длиной волны STED-лазера. Последовательное сканирование всего исследуемого образца дает полную картину со сверхразрешением. Другими словами, STED-микроскоп — это лазерный сканирующий конфокальный микроскоп с дополнительным STED-лазером, субдифракционное разрешение в котором достигается путем избирательного тушения флюорофора [56].
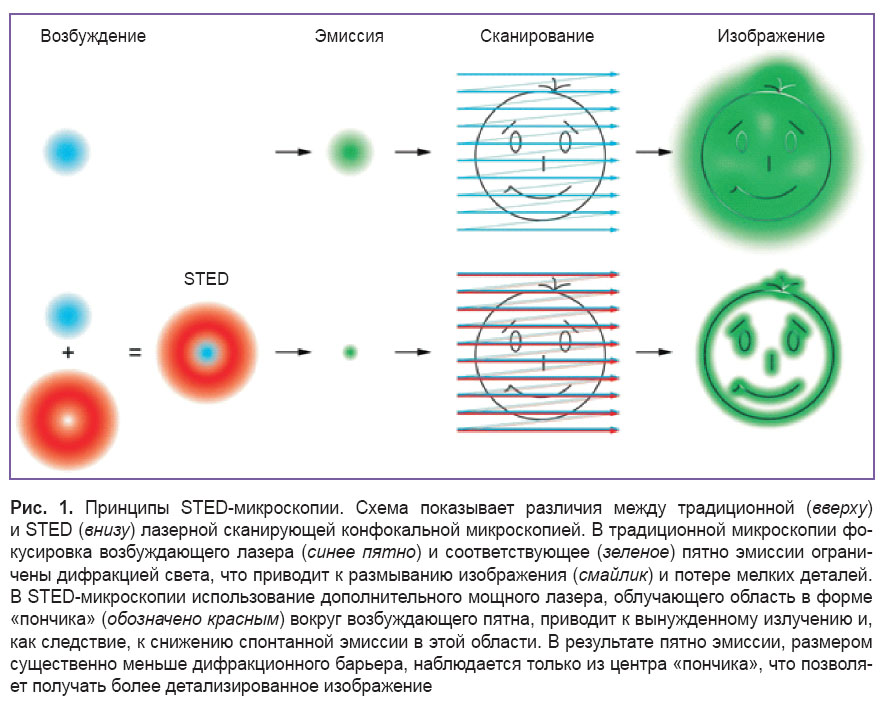
Чем выше мощность STED-лазера, тем лучшее разрешение можно получить. Теоретически можно достичь сколь угодно высокого разрешения [57], однако на практике фотоповреждение биологического образца ведет к ограничению мощности STED-пучка и разрешающая способность обычно составляет примерно 30–80 нм в плоскости xy [8].
Использование времяразрешенных детекторов, синхронизированных с импульсным возбуждающим лазером (gated STED), позволяет существенно снизить мощность STED-лазера. В подобных системах помимо интенсивности флюоресценции учитывается время жизни флюоресцентного состояния, что обеспечивает отбрасывание фотонов с временем жизни флюоресценции, сильно отличающимся от ожидаемого. Это позволяет достигать сверхразрешения и высокого соотношения сигнал/шум в изображении при пониженной интенсивности STED-лазера, снижая риск фотоповреждения живых образцов [58].
Системы на базе STED-микроскопа оказались более популярны, чем методы микроскопии ближнего поля, и нашли широкое применение при решении как физических [59], так и биологических задач, особенно в области нейробиологии. Так, с применением STED-микроскопа были визуализированы ультратонкие структуры дендритных шипиков пирамидальных нейронов в срезах гиппокампа мыши [60]. Детально описаны перемещения везикул, содержащих нейротрансмиттеры, в первичных культурах нейронов крысы [61]. Позднее было продемонстрировано успешное приложение STED-микроскопии для изучения динамики нейронов в коре головного мозга мыши in vivo [62].
Важное преимущество STED-микроскопии состоит в том, что улучшение в разрешении достигается в режиме реального времени и обычно не требует долговременной обработки данных.
Главные недостатки метода — это дороговизна и высокоинтенсивное лазерное облучение, что сильно сужает круг пригодных флюорофоров [63]. С практической точки зрения STED-микроскопия эффективна в основном для работы с фиксированными образцами, меченными фотостабильными флюоресцентными красителями [64]. Получение многоцветных STED-изображений крайне сложно, так как требует тщательно продуманной конструкции детектирующих каналов и комбинации определенных красителей [65].
Принцип, лежащий в основе STED-микроскопии, может быть обобщен и представлен в виде схемы, использующей обратимый переход между двумя состояниями молекулы — флюоресцентным и темновым [66]. Такая концепция воплощена в методе RESOLFT (reversible saturable optical linear fluorescence transitions — микроскопия обратимого насыщенного оптического линейного флюоресцентного перехода). Технология RESOLFT реализована за счет долгоживущих темновых и флюоресцентных состояний обратимо фотопереключаемых флюорофоров с использованием лазеров с особым распределением интенсивности в фокусе аналогично методу STED [67]. Для запуска подобных оптических переходов внутри флюорофора достаточны более низкие мощности лазера, пригодные для исследования живых клеток и тканей с разрешением 50–100 нм [68]. Метод RESOLFT позволил отслеживать динамические перестройки актина в дендритных шипиках живых нейронов гиппокампа в течение нескольких часов без признаков деструкции ткани, с разрешением, в 3 раза превосходящим разрешение конфокального микроскопа [69].
Локализационная микроскопия одиночных молекул (SMLM). В 2006 г. три научные лаборатории независимо друг от друга продемонстрировали новый принцип сверхразрешающей микроскопии. Были представлены такие методы, как STORM (stochastic optical reconstruction microscopy — микроскопия стохастической оптической реконструкции) [70], PALM (photoactivated localization microscopy — фотоактивируемая локализационная микроcкопия) [71] и FPALM (fluorescence photoactivation localization microscopy — флюоресцентная фотоактивируемая локализационная микроскопия) [72], которые положили начало локализационной микроскопии одиночных молекул (SMLM). Главным условием SMLM является детекция флюоресценции одиночных молекул, что позволяет вычислить положение данных молекул с чрезвычайно высокой точностью (рис. 2, а). Для получения данных о множестве молекул необходимо «мигание» флюорофора, т.е. переход между темновым и флюоресцентным состояниями [73]. При этом «включения» и «выключения» флюорофора в отличие от STED и RESOLFT происходят случайным образом, а не в заданных координатах пространства [74, 75]. Детектор микроскопа накапливает сигнал от «включенного» флюорофора, и неопределенность его локализации обратно пропорциональна количеству детектированных фотонов [76].
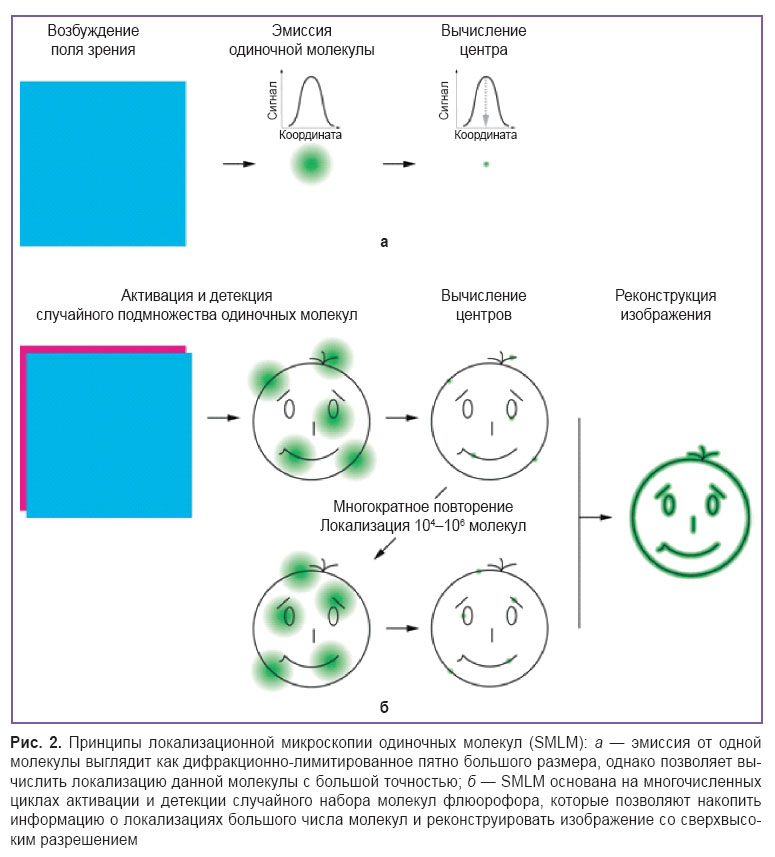 |
Рис. 2. Принципы локализационной микроскопии одиночных молекул (SMLM): а — эмиссия от одной молекулы выглядит как дифракционно-лимитированное пятно большого размера, однако позволяет вычислить локализацию данной молекулы с большой точностью; б — SMLM основана на многочисленных циклах активации и детекции случайного набора молекул флюорофора, которые позволяют накопить информацию о локализациях большого числа молекул и реконструировать изображение со сверхвысоким разрешением |
Физический смысл SMLM заключается в последовательной визуализации подмножеств флюорофоров, рассеянных по тысячам кадров, с целью построения карты реального распределения их плотности (рис. 2, б) [77]. Качество такой реконструкции сильно зависит от алгоритма обработки данных. Разрабатываются различные компьютерные программы для обнаружения и локализации центров точечных источников излучения [78]. Анализ исходных данных обычно включает в себя идентификацию индивидуальных флюорофоров, уточнение координат каждой молекулы флюорофора, реконструкцию высокоразрешенного изображения, на котором обозначены позиции всех локализованных молекул, путем наложения исходных кадров микроскопической съемки друг на друга [79]. Чаще всего разрешающую способность рассчитывают посредством многократного измерения локализации одной и той же молекулы и вычисления стандартного отклонения нормального распределения полученных данных. В качестве альтернативы может быть применен анализ, основанный на преобразованиях Фурье, который учитывает плотность и характер распределения флюорофоров, оценивая сходство двух независимых реконструкций одних и тех же исходных данных [80, 81].
Точность локализации в латеральной плоскости обычно составляет 20–50 нм, в то время как разрешение по оси Z сильно варьирует в зависимости от конфигурации микроскопа [8]. В методах, основанных на использовании двух объективов, расположенных по обе стороны от образца, таких как dual-objective-STORM [82] и iPALM (interferometric-PALM) [83], аксиальное разрешение может достигать 20 нм; при этом стандартный TIRF-режим обеспечивает разрешение по Z в 100 нм. Для получения информации о положении молекулы по оси Z могут также использоваться дополнительные цилиндрические линзы перед детектором, при этом изображение функции рассеяния точки закономерно искажается при удалении от фокуса [84].
Помимо PALM и STORM внедряются новые методики, призванные расширить возможности локализационной микроскопии одиночных молекул, среди которых можно отметить BaLM (bleaching/blinking assisted localization microscopy) [85] и gSHRImP (generalized single-molecule high-resolution imaging with photobleaching) [86] с разрешающей способностью около 30 и 50 нм соответственно. Их принцип основан на извлечении информации о положении каждого флюорофора не только из процесса его мигания, но и фотообесцвечивания, которое, по сути, тоже происходит случайным образом. Такой подход расширяет арсенал пригодных флюоресцентных меток, а также позволяет восстановить картину распределения одиночных молекул при их высокой плотности [87].
Также предложен метод SOFI (super-resolution optical fluctuation imaging), основанный на статистическом анализе временных колебаний флюоресценции флюорофора. SOFI-реконструкция возможна при анализе меньшего числа кадров, чем в методах PALM и STORM, однако требует отсутствия промежутков между кадрами и, как правило, высокой скорости съемки [88]. К преимуществам SOFI относится возможность анализа образцов с высокой плотностью мечения, а также значительное снижение фонового сигнала на результирующем изображении [89].
Среди всех других технологий флюоресцентной микроскопии сверхвысокого разрешения локализационная микроскопия отличается наилучшим соотношением цена/качество. Для использования этого метода на практике достаточно иметь широкопольный микроскоп с набором непрерывных лазеров мощностью около 100 мВт, высокоапертурный объектив и чувствительную CCD-камеру. Для обработки данных в свободном доступе имеется разнообразное программное обеспечение, как, например, rapidSTORM [90] или quickPALM [91]. Более того, ведущие производители оптических приборов Nikon, Zeiss и Leica предлагают исследователям полностью интегрированные системы для SMLM [92].
Заключение
К настоящему моменту методы высокоразрешающей микроскопии остаются новаторскими и сохраняют высокий потенциал применения в различных областях биологии и биомедицины. Постоянно идет поиск и создание эффективных флюорофоров, совершенствуются алгоритмы обработки данных, предлагаются новые схемы организации оптического пути микроскопа [93]. Разработан оригинальный принцип реализации 3D-имиджинга живых клеток [94], апробированы новые подходы к мечению белковых молекул [95] и обработке данных [96]. В последние годы все активнее развиваются технологии, которые объединяют в себе флюоресцентную микроскопию сверхвысокого разрешения с другими методами визуализации. Так, корреляционная световая и электронная микроскопия (correlated light and electron microscopy — CLEM) становится особенно эффективной, когда сочетается с методами на основе PALM, позволяя соотносить электронные микрофотографии с флюоресцентными изображениями в нанометровой шкале [97]. Успешно работает комбинирование высокоразрешающей микроскопии и флюоресцентной микроскопии плоскостного освещения (light sheet fluorescence microscopy — LSFM), позволяющее визуализировать объемные живые биологические образцы в течение длительного времени. Продемонстрирован 3D-имиджинг живых образцов толщиной до 150 мкм с субдифракционным разрешением, основанный на локализации индивидуальных молекул в режиме селективного планарного освещения (individual molecule localization with selective plane illumination microscopy — IML-SPIM) [98]. Разработана технология на базе микроскопии светового листа с дискретным освещением (lattice light sheet microscopy — LLSM) со сверхвысоким разрешением, направленная на визуализацию быстрых динамических процессов на уровне целого организма [99]. Метод LLSM также был успешно совмещен с технологией PAINT, что позволило реализовать многоцветный 3D-имиджинг толстых, с высокой плотностью мечения образцов делящихся клеток и эмбрионов модельных организмов [100].
Одна из основных тенденций — это создание методик, наименее повреждающих живые клетки и пригодных для изучения динамических процессов. Продемонстрированы модифицированные варианты SIM-микроскопии с улучшенным разрешением, работающие на высокой скорости и низких мощностях лазерного облучения (high-numerical aperture TIRF-SIM и patterned activation nonlinear-SIM) [101]. Перспективным представляется взаимодополнение сверхразрешающей микроскопии и технологии микрофлюидики, позволяющее изучать субклеточные процессы в живых объектах с контролируемым 3D-микроокружением, воспроизводящим естественные физиологические условия [102].
Важно подчеркнуть, что развитие подобных технологий возможно только при тесном сотрудничестве ученых различного профиля: инженеров, физиков, программистов, математиков, биофизиков, химиков, молекулярных биологов [103]. Междисциплинарный характер высокоразрешающей микроскопии, с одной стороны, способствует быстрому прогрессу и прорывным достижениям, а с другой — может накладывать ограничение на ее практическое внедрение. Ожидается, что в ближайшие годы технологии высокоразрешающей микроскопии найдут более широкое применение в решении теоретических и прикладных задач и станут рутинной процедурой для исследователей.
Финансирование исследования. Работа выполнена при финансовой поддержке Российского научного фонда (проект №14-25-00129).
Конфликт интересов. У авторов нет конфликта интересов.
Литература
- Periasamy A. Methods in Cellular Imaging. Springer; 2001.
- Ishikawa-Ankerhold H.C., Ankerhold R., Drummen G.P.C. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules 2012; 17(4): 4047–4132, http://dx.doi.org/10.3390/molecules17044047.
- Ettinger A., Wittmann T. Fluorescence live cell imaging. Methods in Cell Biology, vol. 123, 2014, http://dx.doi.org/10.1016/b978-0-12-420138-5.00005-7.
- Lippincott-Schwartz J., Patterson G.H. Development and Use of Fluorescent Protein Markers in Living Cells. Science 2003; 300(5616): 87–91, http://dx.doi.org/10.1126/science.1082520.
- Chudakov D.M., Matz M.V., Lukyanov S., Lukyanov K.A. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol Rev 2010; 90(3): 1103–1163, http://dx.doi.org/10.1152/physrev.00038.2009.
- Lichtman J.W., Conchello J.A. Fluorescence microscopy. Nat Methods 2005; 2(12): 910–919, http://dx.doi.org/10.1038/nmeth817.
- Stehbens S., Pemble H., Murrow L., Wittmann T. Imaging intracellular protein dynamics by spinning disk confocal microscopy. Methods Enzymol 2012; 504: 293–313, http://dx.doi.org/10.1016/B978-0-12-391857-4.00015-X.
- Schermelleh L., Heintzmann R., Leonhardt H. A guide to super-resolution fluorescence microscopy. J Cell Biol 2010; 190(2): 165–175, http://dx.doi.org/10.1083/jcb.201002018.
- Gu M. Advanced Optical Imaging Theory. Springer Science & Business Media; 2000.
- Huang B., Bates M., Zhuang X. Super-Resolution Fluorescence Microscopy. Annu Rev Biochem 2009; 78(1): 993–1016, http://dx.doi.org/10.1146/annurev.biochem.77.061906.092014.
- Stelzer E.H.K. Beyond the diffraction limit? Nature 2002; 417(6891): 806–807, http://dx.doi.org/10.1038/417806a.
- Zhang X., Liu Z. Superlenses to overcome the diffraction limit. Nat Mater 2008; 7(6): 435–441, http://dx.doi.org/10.1038/nmat2141.
- Wiedenmann J., Oswald F., Nienhaus G.U. Fluorescent proteins for live cell imaging: Opportunities, limitations, and challenges. IUBMB Life 2009; 61(11): 1029–1042, http://dx.doi.org/10.1002/iub.256.
- Wäldchen S., Lehmann J., Klein T., van de Linde S., Sauer M. Light-induced cell damage in live-cell super-resolution microscopy. Sci Rep 2015; 5: 15348, http://dx.doi.org/10.1038/srep15348.
- Botchway S.W., Scherer K.M., Hook S., Stubbs C.D., Weston E., Bisby R.H., Parker A.W. A series of flexible design adaptations to the Nikon E-C1 and E-C2 confocal microscope systems for UV, multiphoton and FLIM imaging. J Microsc 2015; 258(1): 68–78, http://dx.doi.org/10.1111/jmi.12218.
- Bozzola J.J., Russell L.D. Electron Microscopy: Principles and Techniques for Biologists. Jones & Bartlett Learning; 1999.
- van Heel M., Gowen B., Matadeen R., Orlova E.V., Finn R., Pape T., Cohen D., Stark H., Schmidt R., Schatz M., Patwardhan A. Single-particle electron cryo-microscopy: towards atomic resolution. Q Rev Biophys 2000; 33(4): 307–369.
- Ayache J., Beaunier L., Boumendil J., Ehret G., Laub D. Sample Preparation Handbook for Transmission Electron Microscopy: Methodology. Springer Science & Business Media; 2010.
- Hayat M. Fixation for Electron Microscopy. Elsevier; 2012.
- Henriques R., Griffiths C., Hesper Rego E., Mhlanga M.M. PALM and STORM: unlocking live-cell super-resolution. Biopolymers 2011; 95(5): 322–331, http://dx.doi.org/10.1002/bip.21586.
- Leung B.O., Chou K.C. Review of Super-Resolution Fluorescence Microscopy for Biology. Appl Spectrosc 2011; 65(9): 967–980, http://dx.doi.org/10.1366/11-06398.
- Klein T., Proppert S., Sauer M. Eight years of single-molecule localization microscopy. Histochem Cell Biol 2014; 141(6): 561–575, http://dx.doi.org/10.1007/s00418-014-1184-3.
- Sydor A.M., Czymmek K.J., Puchner E.M., Mennella V. Super-Resolution Microscopy: From Single Molecules to Supramolecular Assemblies. Trends Cell Biol 2015; 25(12): 730–748, http://dx.doi.org/10.1016/j.tcb.2015.10.004.
- Möckl L., Lamb D.C., Bräuchle C. Super-resolved Fluorescence Microscopy: Nobel Prize in Chemistry 2014 for Eric Betzig, Stefan Hell, and William E. Moerner. Angew Chem Int Ed 2014; 53(51): 13972–13977, http://dx.doi.org/10.1002/anie.201410265.
- Stelzer E.H.K. Better Imaging through Chemistry. Cell 2014; 159(6): 1243–1246, http://dx.doi.org/10.1016/j.cell.2014.11.032.
- Betzig E., Trautman J.K. Near-field optics: microscopy, spectroscopy, and surface modification beyond the diffraction limit. Science 1992; 257(5067): 189–195, http://dx.doi.org/10.1126/science.257.5067.189.
- Dunn R.C. Near-field scanning optical microscopy. Chem Rev 1999; 99(10): 2891–2928, http://dx.doi.org/10.1021/cr980130e.
- Alù A., Engheta N. Cloaked Near-Field Scanning Optical Microscope Tip for Noninvasive Near-Field Imaging. Phys Rev Lett 2010; 105(26), http://dx.doi.org/10.1103/physrevlett.105.263906.
- Oshikane Y., Kataoka T., Okuda M., Hara S., Inoue H., Nakano M. Observation of nanostructure by scanning near-field optical microscope with small sphere probe. Sci Technol Adv Mater 2007; 8(3): 181–185, http://dx.doi.org/10.1016/j.stam.2007.02.013.
- Fernández-Suárez M., Ting A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol 2008; 9(12): 929–943, http://dx.doi.org/10.1038/nrm2531.
- Han R., Li Z., Fan Y., Jiang Y. Recent Advances in Super-Resolution Fluorescence Imaging and Its Applications in Biology. J Genet Genomics 2013; 40(12): 583–595, http://dx.doi.org/10.1016/j.jgg.2013.11.003.
- Dickenson N.E., Armendariz K.P., Huckabay H.A., Livanec P.W., Dunn R.C. Near-field scanning optical microscopy: a tool for nanometric exploration of biological membranes. Anal Bioanal Chem 2010; 396(1): 31–43, http://dx.doi.org/10.1007/s00216-009-3040-1.
- van Zanten T.S., Cambi A., Garcia-Parajo M.F. A nanometer scale optical view on the compartmentalization of cell membranes. Biochim Biophys Acta 2010; 1798(4): 777–787, http://dx.doi.org/10.1016/j.bbamem.2009.09.012.
- Yamamura H., Suzuki Y., Imaizumi Y. New light on ion channel imaging by total internal reflection fluorescence (TIRF) microscopy. J Pharmacol Sci 2015; 128(1): 1–7, http://dx.doi.org/10.1016/j.jphs.2015.04.004.
- Fu Y., Winter P.W., Rojas R., Wang V., McAuliffe M., Patterson G.H. Axial superresolution via multiangle TIRF microscopy with sequential imaging and photobleaching. Proc Natl Acad Sci U S A 2016; 113(16): 4368–4373, http://dx.doi.org/10.1073/pnas.1516715113.
- Axelrod D. Total Internal Reflection Fluorescence Microscopy in Cell Biology. Traffic 2001; 2(11): 764–774, http://dx.doi.org/10.1034/j.1600-0854.2001.21104.x.
- Kudalkar E.M., Davis T.N., Asbury C.L. Single-Molecule Total Internal Reflection Fluorescence Microscopy. Cold Spring Harb Protoc 2016; 2016(5): db.top077800, http://dx.doi.org/10.1101/pdb.top077800.
- Jaiswal J.K., Simon S.M. Imaging single events at the cell membrane. Nat Chem Biol 2007; 3(2): 92–98, http://dx.doi.org/10.1038/nchembio855.
- Rappoport J.Z. Focusing on clathrin-mediated endocytosis. Biochem J 2008; 412(3): 415–423, http://dx.doi.org/10.1042/BJ20080474.
- Chung E., Kim D., Cui Y., Kim Y.H., So P.T.C. Two-Dimensional Standing Wave Total Internal Reflection Fluorescence Microscopy: Superresolution Imaging of Single Molecular and Biological Specimens. Biophys J 2007; 93(5): 1747–1757, http://dx.doi.org/10.1529/biophysj.106.097907.
- Eggeling C., Willig K.I., Sahl S.J., Hell S.W. Lens-based fluorescence nanoscopy. Q Rev Biophys 2015; 48(2): 178–243, http://dx.doi.org/10.1017/s0033583514000146.
- Lippincott-Schwartz J., Manley S. Putting super-resolution fluorescence microscopy to work. Nat Methods 2009; 6(1): 21–23, http://dx.doi.org/10.1038/nmeth.f.233.
- Dyba M., Marcus D., Hell S.W. Focal Spots of Size λ / 23 Open Up Far-Field Florescence Microscopy at 33 nm Axial Resolution. Phys Rev Lett 2002; 88(16), http://dx.doi.org/10.1103/physrevlett.88.163901.
- Bewersdorf J., Schmidt R., Hell S.W. Comparison of I5M and 4Pi-microscopy. J Microsc 2006; 222(Pt 2): 105–117, http://dx.doi.org/10.1111/j.1365-2818.2006.01578.x.
- Nienhaus K., Nienhaus G.U. Where Do We Stand with Super-Resolution Optical Microscopy? J Mol Biol 2016; 428(2 Pt A): 308–322, http://dx.doi.org/10.1016/j.jmb.2015.12.020.
- Gustafsson M.G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 2000; 198(Pt 2): 82–87.
- Yamanaka M., Smith N.I., Fujita K. Introduction to super-resolution microscopy. Microscopy 2014; 63(3): 177–192, http://dx.doi.org/10.1093/jmicro/dfu007.
- Komis G., Mistrik M., Šamajová O., Ovečka M., Bartek J., Šamaj J. Superresolution live imaging of plant cells using structured illumination microscopy. Nat Protoc 2015; 10(8): 1248–1263, http://dx.doi.org/10.1038/nprot.2015.083.
- Schermelleh L., Carlton P.M., Haase S., Shao L., Winoto L., Kner P., Burke B., Cardoso M.C., Agard D.A., Gustafsson M.G.L., Leonhardt H., Sedat J.W. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008; 320(5881): 1332–1336, http://dx.doi.org/10.1126/science.1156947.
- Fitzgibbon J., Bell K., King E., Oparka K. Super-resolution imaging of plasmodesmata using three-dimensional structured illumination microscopy. Plant Physiol 2010; 153(4): 1453–1463, http://dx.doi.org/10.1104/pp.110.157941.
- Lakadamyali M. Super-resolution microscopy: going live and going fast. Chemphyschem 2014; 15(4): 630–636, http://dx.doi.org/10.1002/cphc.201300720.
- Gustafsson M.G.L. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A 2005; 102(37): 13081–13086, http://dx.doi.org/10.1073/pnas.0406877102.
- Zhang H., Zhao M., Peng L. Nonlinear structured illumination microscopy by surface plasmon enhanced stimulated emission depletion. Opt Express 2011; 19(24): 24783–24794, http://dx.doi.org/10.1364/OE.19.024783.
- Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 1994; 19(11): 780–782.
- Blom H., Widengren J. STED microscopy — towards broadened use and scope of applications. Curr Opin Chem Biol 2014; 20: 127–133, http://dx.doi.org/10.1016/j.cbpa.2014.06.004.
- Willig K.I., Keller J., Bossi M., Hell S.W. STED microscopy resolves nanoparticle assemblies. New J Phys 2006; 8(6): 106–106, http://dx.doi.org/10.1088/1367-2630/8/6/106.
- Harke B., Keller J., Ullal C.K., Westphal V., Schönle A., Hell S.W. Resolution scaling in STED microscopy. Opt Express 2008; 16(6): 4154–4162, http://dx.doi.org/10.1364/OE.16.004154.
- Vicidomini G., Moneron G., Han K.Y., Westphal V., Ta H., Reuss M., Engelhardt J., Eggeling C., Hell S.W. Sharper low-power STED nanoscopy by time gating. Nat Methods 2011; 8(7): 571–573, http://dx.doi.org/10.1038/nmeth.1624.
- Rittweger E., Han K.Y., Irvine S.E., Eggeling C., Hell S.W. STED microscopy reveals crystal colour centres with nanometric resolution. Nat Photonics 2009; 3(3): 144–147, http://dx.doi.org/10.1038/nphoton.2009.2.
- Nägerl U.V., Willig K.I., Hein B., Hell S.W., Bonhoeffer T. Live-cell imaging of dendritic spines by STED microscopy. Proc Natl Acad Sci U S A 2008; 105(48): 18982–18987, http://dx.doi.org/10.1073/pnas.0810028105.
- Lauterbach M.A., Keller J., Schönle A., Kamin D., Westphal V., Rizzoli S.O., Hell S.W. Comparing video-rate STED nanoscopy and confocal microscopy of living neurons. J Biophotonics 2010; 3(7): 417–424, http://dx.doi.org/10.1002/jbio.201000038.
- Berning S., Willig K.I., Steffens H., Dibaj P., Hell S.W. Nanoscopy in a Living Mouse Brain. Science 2012; 335(6068): 551–551, http://dx.doi.org/10.1126/science.1215369.
- Neupane B., Ligler F.S., Wang G. Review of recent developments in stimulated emission depletion microscopy: applications on cell imaging. J Biomed Opt 2014; 19(8): 080901, http://dx.doi.org/10.1117/1.JBO.19.8.080901.
- Combs C.A. Fluorescence microscopy: a concise guide to current imaging methods. Curr Protoc Neurosci 2010; Chapter 2: Unit2.1, http://dx.doi.org/10.1002/0471142301.ns0201s50.
- Bückers J., Wildanger D., Vicidomini G., Kastrup L., Hell S.W. Simultaneous multi-lifetime multi-color STED imaging for colocalization analyses. Opt Express 2011; 19(4): 3130–3143, http://dx.doi.org/10.1364/OE.19.003130.
- Hell S.W. Toward fluorescence nanoscopy. Nat Biotechnol 2003; 21(11): 1347–1355, http://dx.doi.org/10.1038/nbt895.
- Schwentker M.A., Bock H., Hofmann M., Jakobs S., Bewersdorf J., Eggeling C., Hell S.W. Wide-field subdiffraction RESOLFT microscopy using fluorescent protein photoswitching. Microsc Res Tech 2007; 70(3): 269–280, http://dx.doi.org/10.1002/jemt.20443.
- Hofmann M., Eggeling C., Jakobs S., Hell S.W. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci U S A 2005; 102(49): 17565–17569, http://dx.doi.org/10.1073/pnas.0506010102.
- Testa I., Urban N.T., Jakobs S., Eggeling C., Willig K.I., Hell S.W. Nanoscopy of Living Brain Slices with Low Light Levels. Neuron 2012; 75(6): 992–1000, http://dx.doi.org/10.1016/j.neuron.2012.07.028.
- Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 2006; 3(10): 793–796, http://dx.doi.org/10.1038/nmeth929.
- Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S., Bonifacino J.S., Davidson M.W., Lippincott-Schwartz J., Hess H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006; 313(5793): 1642–1645, http://dx.doi.org/10.1126/science.1127344.
- Hess S.T., Girirajan T.P.K., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J 2006; 91(11): 4258–4272, http://dx.doi.org/10.1529/biophysj.106.091116.
- Sengupta P., Van Engelenburg S., Lippincott-Schwartz J. Visualizing Cell Structure and Function with Point-Localization Superresolution Imaging. Dev Cell 2012; 23(6): 1092–1102, http://dx.doi.org/10.1016/j.devcel.2012.09.022.
- Vogelsang J., Steinhauer C., Forthmann C., Stein I.H., Person-Skegro B., Cordes T., Tinnefeld P. Make them blink: probes for super-resolution microscopy. Chemphyschem 2010; 11(12): 2475–2490, http://dx.doi.org/10.1002/cphc.201000189.
- Tam J., Merino D. Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J Neurochem 2015; 135(4): 643–658, http://dx.doi.org/10.1111/jnc.13257.
- Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys J 2002; 82(5): 2775–2783, http://dx.doi.org/10.1016/S0006-3495(02)75618-X.
- Almada P., Culley S., Henriques R. PALM and STORM: Into large fields and high-throughput microscopy with sCMOS detectors. Methods 2015; 88: 109–121, http://dx.doi.org/10.1016/j.ymeth.2015.06.004.
- Sage D., Kirshner H., Pengo T., Stuurman N., Min J., Manley S., Unser M. Quantitative evaluation of software packages for single-molecule localization microscopy. Nat Methods 2015; 12(8): 717–724, http://dx.doi.org/10.1038/nmeth.3442.
- Li Y., Ishitsuka Y., Hedde P.N., Nienhaus G.U. Fast and efficient molecule detection in localization-based super-resolution microscopy by parallel adaptive histogram equalization. ACS Nano 2013; 7(6): 5207–5214, http://dx.doi.org/10.1021/nn4009388.
- Endesfelder U., Heilemann M. Art and artifacts in single-molecule localization microscopy: beyond attractive images. Nat Methods 2014; 11(3): 235–238, http://dx.doi.org/10.1038/nmeth.2852.
- Banterle N., Bui K.H., Lemke E.A., Beck M. Fourier ring correlation as a resolution criterion for super-resolution microscopy. J Struct Biol 2013; 183(3): 363–367, http://dx.doi.org/10.1016/j.jsb.2013.05.004.
- Xu K., Babcock H.P., Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods 2012; 9(2): 185–188, http://dx.doi.org/10.1038/nmeth.1841.
- Shtengel G., Galbraith J.A., Galbraith C.G., Lippincott-Schwartz J., Gillette J.M., Manley S., Sougrat R., Waterman C.M., Kanchanawong P., Davidson M.W., Fetter R.D., Hess H.F. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci U S A 2009; 106(9): 3125–3130, http://dx.doi.org/10.1073/pnas.0813131106.
- Huang B., Wang W., Bates M., Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008; 319(5864): 810–813, http://dx.doi.org/10.1126/science.1153529.
- Burnette D.T., Sengupta P., Dai Y., Lippincott-Schwartz J., Kachar B. Bleaching/blinking assisted localization microscopy for superresolution imaging using standard fluorescent molecules. Proc Natl Acad Sci U S A 2011; 108(52): 21081–21086, http://dx.doi.org/10.1073/pnas.1117430109.
- Simonson P.D., Rothenberg E., Selvin P.R. Correction to Single-Molecule-Based Super-Resolution Images in the Presence of Multiple Fluorophores. Nano Lett 2013; 13(3): 1366–1366, http://dx.doi.org/10.1021/nl4004267.
- Wang Y., Kuang C., Cai H., Li S., Liu W., Hao X., Ge J., Liu X. Sub-diffraction imaging with confocal fluorescence microscopy by stochastic photobleaching. Opt Commun 2014; 312: 62–67, http://dx.doi.org/10.1016/j.optcom.2013.09.022.
- Dertinger T., Colyer R., Iyer G., Weiss S., Enderlein J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc Natl Acad Sci U S A 2009; 106(52): 22287–22292, http://dx.doi.org/10.1073/pnas.0907866106.
- Geissbuehler S., Dellagiacoma C., Lasser T. Comparison between SOFI and STORM. Biomed Opt Express 2011; 2(3): 408–420, http://dx.doi.org/10.1364/BOE.2.000408.
- Wolter S., Löschberger A., Holm T., Aufmkolk S., Dabauvalle M.C., van de Linde S., Sauer M. rapidSTORM: accurate, fast open-source software for localization microscopy. Nat Methods 2012; 9(11): 1040–1041, http://dx.doi.org/10.1038/nmeth.2224.
- Henriques R., Lelek M., Fornasiero E.F., Valtorta F., Zimmer C., Mhlanga M.M. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat Methods 2010; 7(5): 339–340, http://dx.doi.org/10.1038/nmeth0510-339.
- Sauer M. Localization microscopy coming of age: from concepts to biological impact. J Cell Sci 2013; 126(Pt 16): 3505–3513, http://dx.doi.org/10.1242/jcs.123612.
- Cox S., Jones G.E. Imaging cells at the nanoscale. Int J Biochem Cell Biol 2013; 45(8): 1669–1678, http://dx.doi.org/10.1016/j.biocel.2013.05.010.
- Geissbuehler S., Sharipov A., Godinat A., Bocchio N.L., Sandoz P.A., Huss A., Jensen N.A., Jakobs S., Enderlein J., Gisou van der Goot F., Dubikovskaya E.A., Lasser T., Leutenegger M. Live-cell multiplane three-dimensional super-resolution optical fluctuation imaging. Nat Commun 2014; 5: 5830, http://dx.doi.org/10.1038/ncomms6830.
- Ries J., Kaplan C., Platonova E., Eghlidi H., Ewers H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods 2012; 9(6): 582–584, http://dx.doi.org/10.1038/nmeth.1991.
- Levet F., Hosy E., Kechkar A., Butler C., Beghin A., Choquet D., Sibarita J.B. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat Methods 2015; 12(11): 1065–1071, http://dx.doi.org/10.1038/nmeth.3579.
- de Souza N. Super-resolution CLEM. Nat Methods 2014; 12(1): 37–37, http://dx.doi.org/10.1038/nmeth.3239.
- Zanacchi F.C., Lavagnino Z., Donnorso M.P., Del Bue A., Furia L., Faretta M., Diaspro A. Live-cell 3D super-resolution imaging in thick biological samples. Nat Methods 2011; 8(12): 1047–1049, http://dx.doi.org/10.1038/nmeth.1744.
- Chen B.C., Legant W.R., Wang K., Shao L., Milkie D.E., Davidson M.W., Janetopoulos C., Wu X.S., Hammer J.A. 3rd, Liu Z., English B.P., Mimori-Kiyosue Y., Romero D.P., Ritter A.T., Lippincott-Schwartz J., Fritz-Laylin L., Mullins R.D., Mitchell D.M., Bembenek J.N., Reymann A.C., Böhme R., Grill S.W., Wang J.T., Seydoux G., Tulu U.S., Kiehart D.P., Betzig E. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 2014; 346(6208): 1257998, http://dx.doi.org/10.1126/science.1257998.
- Legant W.R., Shao L., Grimm J.B., Brown T.A., Milkie D.E., Avants B.B., Lavis L.D., Betzig E. High-density three-dimensional localization microscopy across large volumes. Nat Methods 2016; 13(4): 359–365, http://dx.doi.org/10.1038/nmeth.3797.
- Li D., Shao L., Chen B.C., Zhang X., Zhang M., Moses B., Milkie D.E., Beach J.R., Hammer J.A. 3rd, Pasham M., Kirchhausen T., Baird M.A., Davidson M.W., Xu P., Betzig E. ADVANCED IMAGING. Extended-resolution structured illumination imaging of endocytic and cytoskeletal dynamics. Science 2015; 349(6251): aab3500, http://dx.doi.org/10.1126/science.aab3500.
- Chin L.K., Lee C.H., Chen B.C. Imaging live cells at high spatiotemporal resolution for lab-on-a-chip applications. Lab Chip 2016, http://dx.doi.org/10.1039/c5lc01556a.
- Whelan D.R., Bell T.D.M. Super-Resolution Single-Molecule Localization Microscopy: Tricks of the Trade. J Phys Chem Lett 2015; 6(3): 374–382, http://dx.doi.org/10.1021/jz5019702.








