Регуляция ферроптоза макрофагов человека донорами оксида азота
Ферроптоз — программируемая форма клеточной гибели, в которой основным звеном является железозависимое перекисное окисление липидов. Макрофаги — основные клетки иммунной системы, они функционируют в проокислительной среде, поэтому изучение их предрасположенности к ферроптозу и поиск подходов к его регуляции имеют важное значение.
Цель исследования — изучить особенности ферроптоза макрофагов, дифференцированных из клеток миелоидной лейкемии ТНР-1, и сравнить влияние доноров NO с разным временем полураспада на степень развития ферроптоза.
Материалы и методы. Для индукции ферроптоза ТНР-1-макрофагов были использованы ингибиторы глутатионпероксидазы 4 (GPX4) — RSL3 и ML-162, а также ингибитор цистин-глутаматного обмена эрастин. За развитием ферроптоза следили с помощью трех независимых методов: восстановление живыми клетками аламарового синего, измерение лактатдегидрогеназы в среде, LIVE/DEAD-тест. Ферроптотическая гибель клеток была доказана с помощью специфического ингибитора ферростатина-1, а также путем выявления окисления липидов в клетках с использованием флуоресцентного зонда BODIPY 581/591 С11.
Результаты. RSL3 и ML-162 дозозависимо индуцировали ферроптоз клеток. Ферроптоз ТНР-1-макрофагов — медленный процесс, он начинается через ~5 ч после добавления индуктора. Эрастин является слабым индуктором ферроптоза, однако он усиливал ферроптоз, вызванный ингибиторами GPX4. Мы сравнили способность двух доноров NO с разным временем полураспада влиять на ферроптоз ТНР-1-макрофагов: DEA NONOate (2 мин) и DPTA NONOate (3 ч). Доноры добавляли однократно после индуктора в концентрации 100–120 мкМ или несколько раз до достижения заданной концентрации. DEA не влиял на ферроптоз ТНР-1-макрофагов, тогда как DPTA полностью ингибировал ферроптоз.
Заключение. DPTA — донор NO cо временем полураспада 3 ч при 37°С — может быть использован для ингибирования ферроптоза ТНР-1-макрофагов, который развивается в течение 17–19 ч. Следовательно, существуют механизмы пролонгирования действия NO, которые необходимо изучать с целью использования доноров NO для регуляции ферроптоза клеток.
Введение
Ферроптоз — это особый тип регулируемой программируемой гибели клеток в результате нарушения трех метаболических процессов: концентрации глутатиона в клетке, метаболизма железа и регуляции перекисного окисления липидов [1, 2]. Перекисное окисление липидов было признано фундаментальным звеном ферроптоза [3, 4]. В настоящее время хорошо известна связь ферроптоза с различными патологиями: начиная от нейродегенеративных заболеваний и повреждений, вызванных ишемией–реперфузией, до заболеваний почек и резистентных видов рака [5–7]. Это стимулирует поиск новых механизмов изменения устойчивости клеток к ферроптозу.
Глутатионпероксидаза 4 (GPX4) катализирует восстановление гидроперекисей липидов (PLOOH) в соответствующие гидроксильные производные (PLOH) за счет окисления тиоловых групп глутатиона (GSH) [8]. Ингибирование GPX4 специфическими ингибиторами RSL3 или ML-162 является стандартной экспериментальной моделью для изучения ферроптоза. В качестве альтернативного индуктора ферроптоза в лабораторных исследованиях широко используют эрастин — ингибитор цистин-глутаматной антипортерной системы Хc–.
Макрофаги — основные клетки врожденного иммунитета, которые участвуют во всех стадиях иммунного ответа при повреждении тканей или воздействиях патогенов, а также координируют действия других клеток. Они продуцируют провоспалительные цитокины и активные формы кислорода, создавая проокислительную среду, которая может способствовать развитию ферроптоза в клетках [9–11]. ТНР-1 представляет собой моноцитарную линию лейкоза человека, активация этих клеток форбол-12-миристат-13-ацетатом приводит к их дифференцировке в макрофагоподобные клетки [12]. Данная клеточная линия является распространенной моделью изучения иммуномодулирующих свойств моноцитов/макрофагов как в области фундаментальной иммунологии, так и в качестве in vitro модели различных заболеваний, в патогенез которых вовлечены макрофаги (например, сердечно-сосудистых заболеваний, нейровоспаления, рака и инфекционных заболеваний) [13–15]. Несмотря на то, что бóльшая часть исследований механизмов ферроптоза макрофагов проводилось на мышиных макрофагах — клеточной линии RAW 264.7 и макрофагах, полученных из костного мозга, в последние годы THP-1 также активно изучаются в контексте заболеваний, где клеточная гибель происходит по ферроптоз-зависимым механизмам. Повышенная концентрация железа при эндометриозе индуцирует ферроптоз у THP-1-макрофагов, что приводит к снижению фагоцитирующей способности этих клеток, а также вызывает усиление секреции ангиогенных цитокинов, таких как фактор роста эндотелия сосудов A и интерлейкин 8. Данные факторы способствуют усилению эндометриоза [16]. Ингибирование ферроптоза у THP-1-макрофагов с помощью байкалеина, который является потенциальным средством против ферроптоза благодаря способности повышать экспрессию GPX4, восстанавливало фагоцитирующую активность клеток. Данное соединение рассматривается как перспективный препарат для терапии эндометриоза посредством ослабления ферроптоза макрофагов [16]. Исходя из этого, можно утверждать, что THP-1-макрофаги представляют собой важную модель для изучения ферроптоз-зависимой клеточной гибели, а также фармакологических соединений, нацеленных на модуляцию процесса ферроптоза в макрофагах.
GSH/GPX4 является преобладающей системой детоксикации гидропероксидов фосфолипидов в клетках млекопитающих [8]. Однако недавно были обнаружены независимые от GPX4 регуляторы ферроптоза, такие как белок-супрессор ферроптоза 1 [17] и Ca2+-независимая фосфолипаза A2β [18]. Ряд природных или синтетических перехватчиков свободных радикалов — от членов семейства витаминов Е до разнообразных ароматических аминов и фенольных соединений — действуют как ингибиторы ферроптоза [19]. Показано, что клетки макрофагов мышей RAW 264.7 способны стимулировать антиферроптотический механизм, управляемый индуцированной NO-синтазой (iNOS), для того чтобы заблокировать перекисное окисление липидов и защитить клетки от гибели [20]. Причем макрофаги могут защитить не только себя, но и клетки легочного эпителия в экспериментах по кокультивированию [21].
Известно, что NO• представляет собой активную молекулу, продуцируемую семейством белков iNOS [22]. Во-первых, NO• является мощной сигнальной молекулой, способной действовать паракринно во время расслабления гладких мышц или расширения сосудов, а также как внутриклеточный вторичный мессенджер, регулирующий антиоксидантный ответ клеток [23]. NO• напрямую связывает и инактивирует Fe-содержащие ферменты и может реагировать с супероксидным анион-радикалом с образованием высокореакционного пероксинитрита, который участвует в защите от патогенов [24]. Кроме того, способность NO• взаимодействовать с различными радикалами (включая промежуточные липидные радикалы), появляющимися в процессе ферроптоза, может облегчить посттрансляционное нитрозилирование белков и липидов, модифицирующее их активность, стабильность или локализацию. Доноры NO — вещества стабильные в органических растворителях или при сильнощелочных рН, в которых готовятся их стоковые растворы. Однако, попадая в среду с рН=7,4, они диспропорционируют с высвобождением газообразного NO.
В настоящей работе мы изучили ферроптоз ТНР-1-макрофагов и показали возможность его регулирования: усиление за счет одновременного действия индукторов разной природы и ингибирование донором оксида азота.
Материалы и методы
Культивирование клеток. Клетки моноцитов человека THP-1 культивировали в среде RPMI 1640 без L-глутамата, с 2 мМ Glutamax, 10% термически инактивированной фетальной бычьей сывороткой (FBS), пенициллином (100 Ед./мл) и стрептомицином (100 мкг/мл) при 37°С и 5% СО2. Дифференцировку в макрофаги осуществляли путем инкубации в течение 48 ч с форбол-12-миристат-13-ацетaтом, добавленным в количестве 75 нг/мл. Через 2 дня среду заменяли и клетки культивировали без форбола-12-миристата-13-ацетaта еще сутки, после чего делали добавки к клеточной среде. Для экспериментов по определению жизнеспособности клеток их высаживали в 48-луночный планшет по 150 тыс. клеток на 1 лунку в 350 мл среды. В качестве индукторов ферроптоза использовали ингибиторы GPX4 — RSL3 и ML-162, которые добавляли в среду инкубации в концентрации 0,5–2,5 мкM и инкубировали в течение 5–22 ч с последующим анализом гибели клеток. Кроме того, для инициации гибели клеток использовали блокатор цистин-глутаматной антипортерной системы эрастин. Для доказательства ферроптотической формы гибели клеток эксперименты проводили с добавлением в среду культивирования ингибитора ферроптоза ферростатина-1 (Fer-1), который предотвращает образование гидроперекисей липидов, в дозе 5 мкM. В экспериментах использовали доноры: Diethylamine NONOate sodium salt hydrate (DEA NONOate) — NO-донор с периодом полураспада 2 мин при 37°C и 15 мин при 22–25°C и Dipropylenetriamine NONOate (DPTA NONOate) — NO-донор с периодом полураспада 3 ч при 37°C и 5 ч при 22–25°C (0,1 M фосфатный буфер, pH=7,4). Первую добавку делали в течение 5–15 мин после добавления индуктора ферроптоза, далее добавляли реагент через определенные промежутки времени.
Характеристика клеточной гибели. Клеточную гибель оценивали количественно с помощью трех независимых методов:
1. Определение лактатдегидрогеназы (ЛДГ) в среде клеток проводили с помощью стандартных наборов детекции ЛДГ (Thermo Fisher Scientific, США). К 50 мкл среды (контроль — среда без инкубации с макрофагами) добавляли 50 мкл реакционного буфера, инкубировали 30 мин. После добавления 50 мкл стоп-реагента измеряли поглощение раствора при 490 и 680 нм. Процент гибели клеток рассчитывали как (ЛДГпроба/ЛДГконтроль)·100%.
2. Аламаровый синий добавляли к клеткам в концентрации 10 мкг/мл; инкубирование проводили при температуре 37°С в течение 2 ч с последующим измерением флуоресценции на спектрофлуориметре VICTOR Nivo (PerkinElmer, США), Ex — 580/20 нм, Em — 625/20 нм.
3. LIVE/DEAD Viability/Cytotoxicity Assay Kit (Thermo Fisher Scientific, США) — стандартный тест для характеристики жизнеспособности клеток, который позволяет отличить живые клетки от мертвых путем одновременного окрашивания зеленым кальцеином AM и красным флуоресцентным гомодимером-1 пропидий йодида. После замены среды к клеткам добавляли реагенты LIVE/DEAD-теста и 1 мкМ Hoechst для окрашивания ядер. Окрашивание проводили в течение 30 мин при температуре 37°C и 5% CO2 согласно инструкции производителя, после чего меняли среду. Живые и мертвые клетки визуализировали с помощью флуоресцентного микроскопа EVOS M5000 (Thermo Fisher Scientific, США) с установленным диапазоном длин волн для возбуждения и регистрации флуоресценции красителей: для Hoechst 33342 (Exmax — 351 нм, Emmax — 461 нм) использовали канал DAPI (возбуждение при длине волны 357±44 нм; излучение регистрируется в диапазоне 447±30 нм); для кальцеина АМ (Exmax — 494 нм, Emmax — 517 нм) использовали канал GFP (возбуждение при длине волны 470±22 нм; излучение регистрируется в диапазоне 525±25 нм); для пропидий йодида (Exmax — 535 нм, Emmax — 617 нм) использовали канал RFP (возбуждение при длине волны 531±40 нм, излучение регистрируется в диапазоне 593±20 нм).
Визуализация окисления липидов клеток с помощью маркера окисления липидов BODIPY. THP-1-клетки сеяли в 48-луночный планшет, через сутки после замены среды к клеткам добавляли зонд BODIPY 581/591 C11 (Invitrogen, США) в концентрации 15 мкМ и инкубировали при 37°С и 5% СО2, спустя 30 мин к экспериментальным клеткам добавляли индуктор ферроптоза RSL3 в концентрации 1,25 мкM. Через 17–19 ч меняли среду, после чего образцы анализировали, используя EVOS M5000. Зонд, встраиваясь в мембрану клеток, имеет флуоресценцию в красной области (Exmax — 581, Emmax — 591, канал GFP), но при окислении параметры флуоресценции молекулы сдвигаются в зеленую область (Exmax — 488, Emmax — 510; канал RFP).
Статистика. Статистический анализ проводили с помощью программы GraphPad Prism 9.0.5. Порог значимости был установлен на уровне р<0,05. Группы сравнивали с использованием двуфакторного дисперсионного анализа ANOVA (two-way ANOVA) и однофакторного анализа ANOVA c пост-хок тестом Тьюки.
Результаты
Индукторы ферроптоза RSL3 или ML-162 были добавлены в среду культивирования ТНР-1-макрофагов в разных концентрациях — 0,5; 1,25 и 2,5 мкМ. За нативностью клеток следили при помощи светового микроскопа, а также ряда биохимических методов: 1) наиболее точным параметром, который позволяет количественно охарактеризовать гибель клеток, является концентрация фермента ЛДГ, вышедшего из мертвых клеток в среду инкубации; 2) простой и наглядный метод на основе аламарового синего позволяет оценить метаболическую активность живых клеток, которые способны восстанавливать аламаровый синий (резазурин) до флуоресцирующего резоруфина; 3) LIVE/DEAD-тест на основе двух красителей позволяет визуально различить живые и мертвые клетки.
Время развития ферроптоза и количество погибших клеток зависят от концентрации индукторов ферроптоза и времени инкубации. На рис. 1 показано, что количество ЛДГ в среде ТНР-1-макрофагов повышалось с увеличением концентрации индуктора ферроптоза и времени инкубации клеток с индуктором. При наших экспериментальных условиях гибель клеток начинается через 5–9 ч инкубации и достигает максимума через 13–19 ч в зависимости от концентрации индуктора.
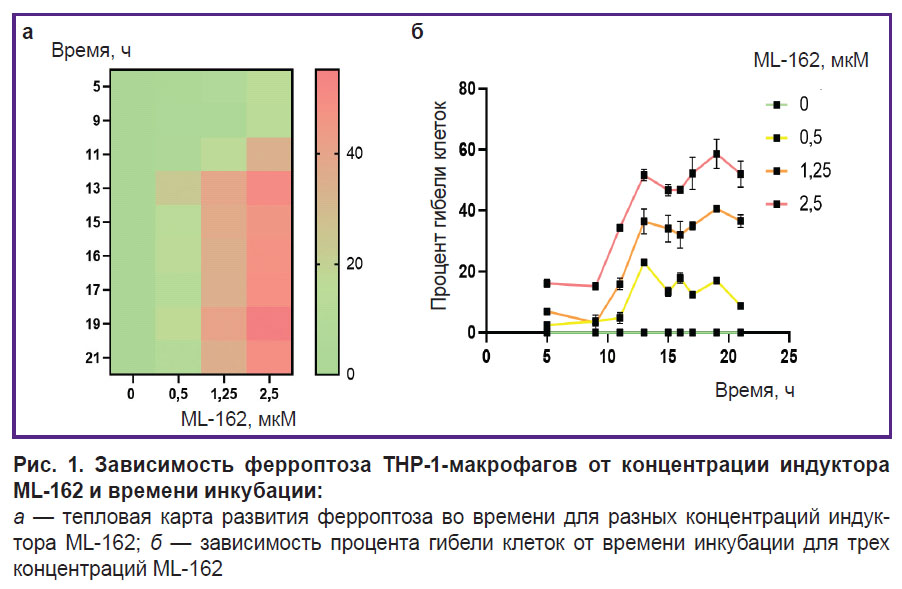
|
Рис. 1. Зависимость ферроптоза ТНР-1-макрофагов от концентрации индуктора ML-162 и времени инкубации: а — тепловая карта развития ферроптоза во времени для разных концентраций индуктора ML-162; б — зависимость процента гибели клеток от времени инкубации для трех концентраций ML-162 |
На рис. 2 показаны результаты эксперимента, в котором сравнивали гибель клеток, измеренную с помощью ЛДГ и аламарового синего. Увеличение концентрации ЛДГ сопровождалось уменьшением количества живых клеток, способных восстановить резазурин. Для приведенного эксперимента наблюдалось хорошее соответствие результатов, полученных двумя независимыми методами. Однако в ряде экспериментов результаты с аламаровым синим не соответствовали реальности (результаты не представлены), что может быть обусловлено окислительной активностью макрофагов или их переходом в среду вследствие инициации гибели. В связи с этим в качестве основного метода количественной характеристики перекисного окисления липидов был выбран метод на основе LDН.
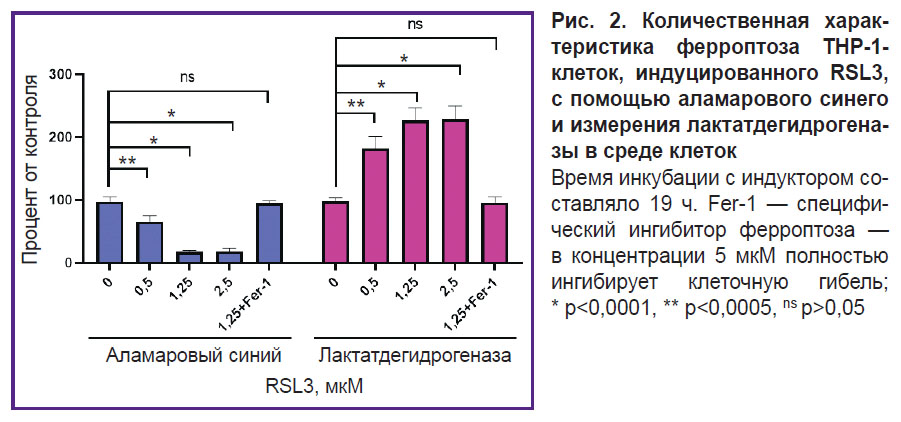
|
Рис. 2. Количественная характеристика ферроптоза ТНР-1-клеток, индуцированного RSL3, с помощью аламарового синего и измерения лактатдегидрогеназы в среде клеток
Время инкубации с индуктором составляло 19 ч. Fer-1 — специфический ингибитор ферроптоза — в концентрации 5 мкМ полностью ингибирует клеточную гибель; * p<0,0001, ** p<0,0005, ns p>0,05 |
Визуализацию клеточной гибели проводили с использованием LIVE/DEAD-теста (рис. 3). В контроле клетки были окрашены в зеленый цвет; живые клетки восстанавливают кальцеин АМ до флуоресцирующего кальцеина. Добавление индуктора ферроптоза RSL3 дозозависимо приводит к уменьшению количества живых клеток и к росту числа клеток, окрашенных пропидий иодидом, который может проникать только в мертвые клетки.
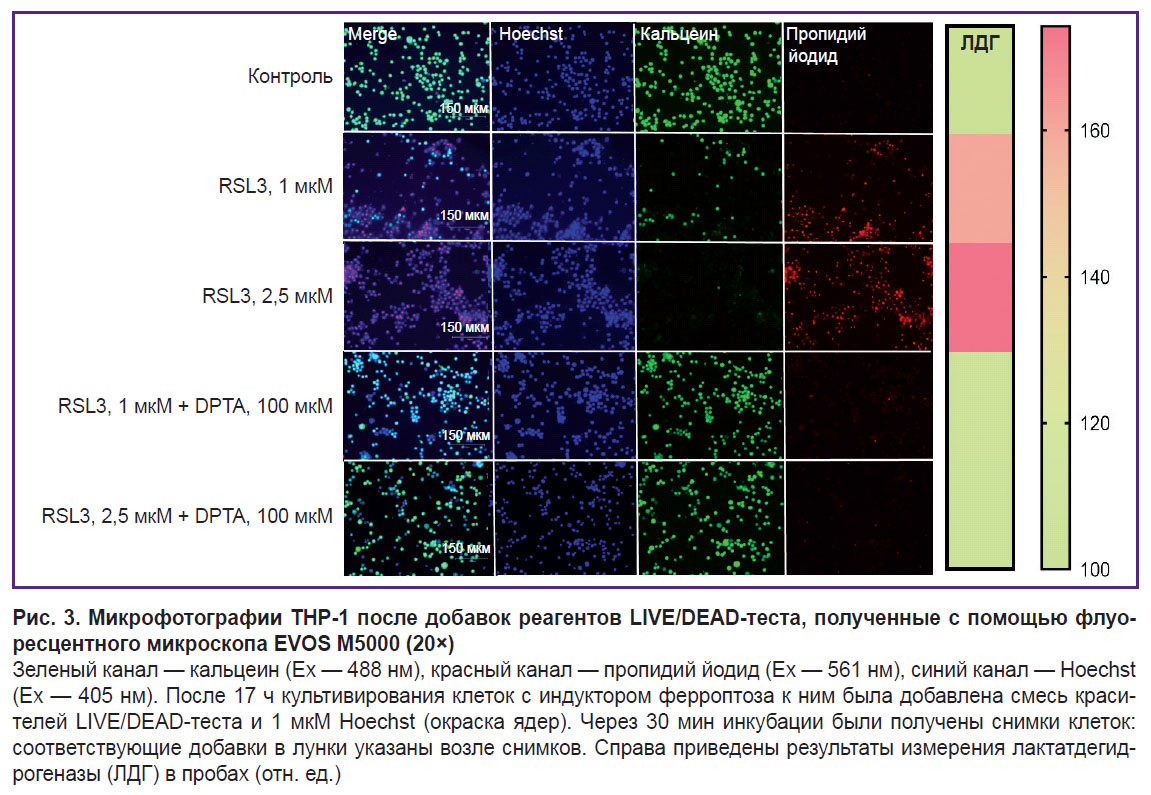
Для доказательства ферроптотической формы гибели клеток мы использовали специфический ингибитор ферроптоза Fer-1 (5 мкМ), который полностью ингибировал клеточную гибель (см. рис. 2). Окисление липидов мембраны клетки — ключевое звено ферроптоза. Чтобы дополнительно доказать, что изучаемая нами гибель клеток является ферроптозом, мы применяли специфический флуоресцентный маркер окисления липидов BODIPY С11 (рис. 4). В наших экспериментах наблюдалось дозозависимое изменение флуоресценции BODIPY: уменьшение флуоресценции в красной области и увеличение в зеленой при повышении концентрации RSL3.
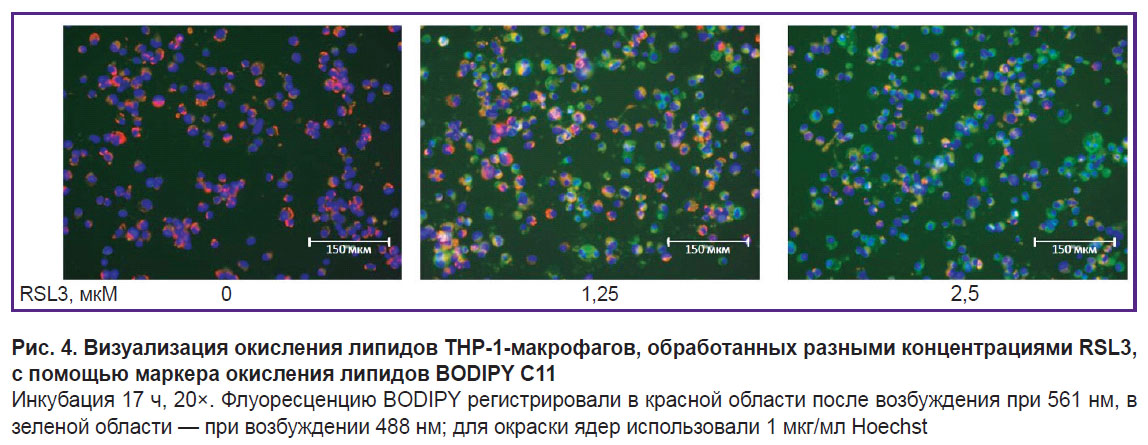
|
Рис. 4. Визуализация окисления липидов ТНР-1-макрофагов, обработанных разными концентрациями RSL3, с помощью маркера окисления липидов BODIPYС11 Инкубация 17 ч, 20×. Флуоресценцию BODIPY регистрировали в красной области после возбуждения при 561 нм, в зеленой области — при возбуждении 488 нм; для окраски ядер использовали 1 мкг/мл Hoechst |
Эрастин слабо влиял на жизнеспособность THP-1-макрофагов, гибель клеток при добавлении 10 мкM эрастина составила 23±7% от контроля. Если клетки одновременно были обработаны индукторами ферроптоза с разными механизмами действия, наблюдалось усиление их гибели, которое превышает суммарное действие используемых индукторов (рис. 5).
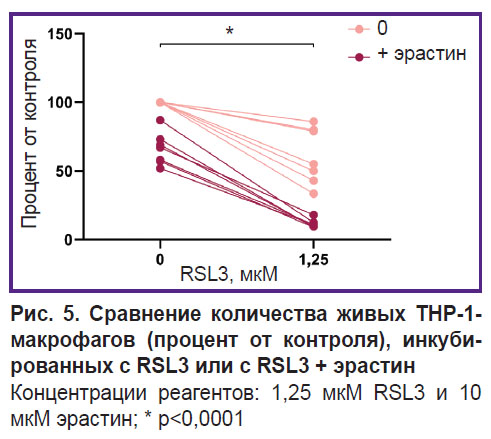
|
Рис. 5. Сравнение количества живых ТНР-1-макрофагов (процент от контроля), инкубированных с RSL3 или с RSL3 + эрастин Концентрации реагентов: 1,25 мкМ RSL3 и 10 мкМ эрастин; * p<0,0001 |
В качестве донора оксида азота применяли DPTA NONOate. DPTA полностью ингибирует ферроптоз ТНР-1-клеток в концентрации 100 мкМ. Ингибирование не зависело от того, сделана ли указанная добавка один раз или 2 раза по 50 мкМ (рис. 6, a). Влияние донора NO на ферроптоз ТНР-1-клеток показано также с помощью LIVE/DEAD-теста (см. рис. 3). Присутствие в среде 100 мкM DPTA полностью ингибирует ферроптоз даже при максимальной концентрации RSL3 (2,5 мкМ): на снимках нет клеток, в которые проникал пропидий иодид.
В экспериментах был также использован донор DEA NONOate. Добавки донора делали после индуктора ферроптоза 2–3 раза по 40 мкМ с интервалом 3 ч. DEA NONOate не влиял на развитие ферроптоза ни при какой концентрации индуктора (рис. 6, б).
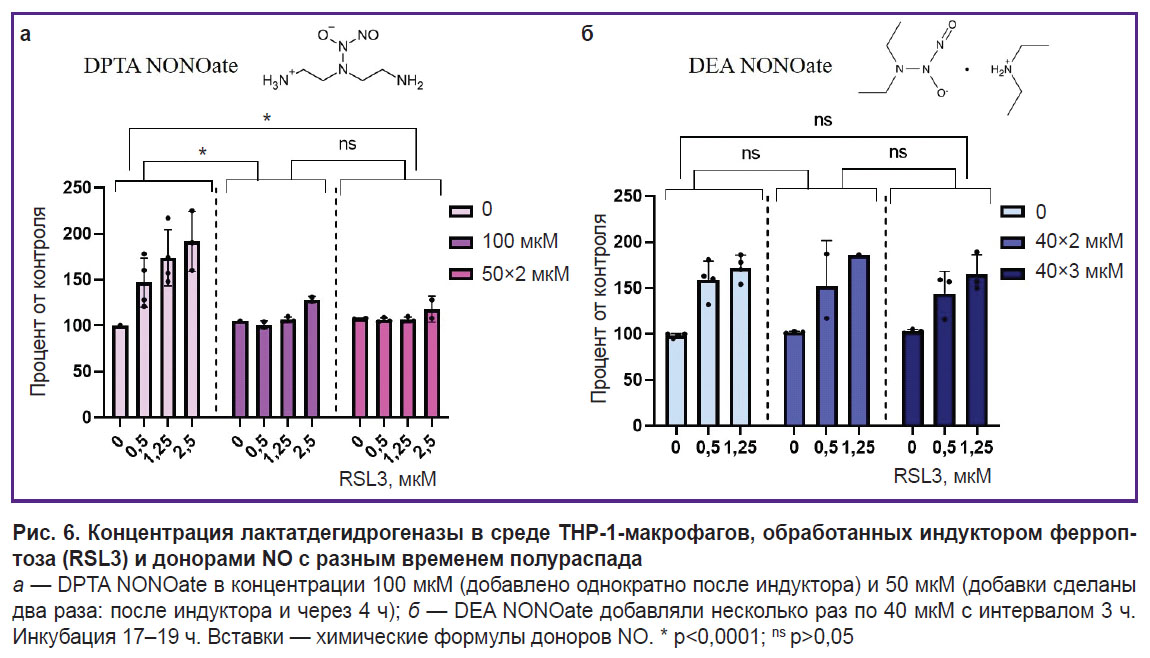
|
Рис. 6. Концентрация лактатдегидрогеназы в среде ТНР-1-макрофагов, обработанных индуктором ферроптоза (RSL3) и донорами NOс разным временем полураспада
а — DPTA NONOate в концентрации 100 мкM (добавлено однократно после индуктора) и 50 мкМ (добавки сделаны два раза: после индуктора и через 4 ч); б — DEA NONOate добавляли несколько раз по 40 мкM c интервалом 3 ч. Инкубация 17–19 ч. Вставки — химические формулы доноров NO. * p<0,0001; ns p>0,05 |
Обсуждение
Ферроптоз — это неапоптотический процесс гибели клеток, фундаментальным звеном которого является Fe-зависимое накопление гидроперекисей липидов. Ферроптотическая гибель клеток лежит в основе ряда заболеваний, таких как нейродегенеративные заболевания, поражения печени и сердца при ишемии–реоксигенации и др. [5, 25]. В связи с этим поиск ингибиторов ферроптоза представляется важной задачей, решение которой поможет в терапии ферроптоз-зависимых патологий [16, 26].
Ферроптотическая форма клеточной гибели показана для многих типов клеток, однако для макрофагов человека эта форма гибели изучена мало. Исследования выполнены в основном для макрофагов мышей [9, 20]. В настоящей работе мы использовали ТНР-1-макрофаги как модель макрофагов человека [27]. Ферроптоз ТНР-1-макрофагов — медленный процесс, он начинает развиваться через ~5 ч после добавки индукторов RSL3 или ML-162. Степень развития ферроптоза зависит от дозы добавленного индуктора и достигает максимума через 17–19 ч после инициации программы смерти (см. рис. 1). Развитие ферроптоза характеризовали тремя независимыми методами: измерение ЛДГ в кондиционированной среде клеток, восстановление аламарового синего до резоруфина живыми клетками и стандартный LIVE-DEAD-тест c кальцеином АМ и пропидий иодидом (см. рис. 2, 3). Результаты разных методов хорошо совпадали, но наиболее простым и достоверным является измерение ЛДГ. Специфический ингибитор ферроптоза Fer-1 полностью ингибирует действие индукторов ферроптоза в концентрации 5 мкМ. Дополнительно доказательство ферроптотической формы гибели клеток было получено с помощью флуоресцентного маркера окисления липидов BODIPY С11. В мембране клеток BODIPY флуоресцирует в красной области, но если проходит массовое окисление липидов, липидные радикалы окисляются BODIPY, что сопровождается сдвигом флуоресценции в зеленую область в клетках, обработанных индуктором ферроптоза. Количество клеток, несущих на себе маркер с зеленой окраской, зависело от дозы индуктора, добавленного в среду инкубации (см. рис. 4).
Результаты для двух индукторов ферроптоза — RSL3 и ML-162, которые являются ингибиторами GPX4, не отличались; индукция ферроптоза ТНР-1-клеток наблюдалась уже при концентрации реагентов 0,5 мкМ. Ингибитор цистин-глутаматного обмена эрастин, который уменьшает концентрацию GSH в клетках, является слабым индуктором ферроптоза в случае ТНР-1-макрофагов, в отличие от ингибиторов GPX4. Это также было показано ранее для макрофагов RAW 264.7 [20]. Гибель клеток при добавлении 10 мкM эрастина составляла 23±11% от контроля. Однако одновременное добавление двух типов индукторов приводило к усилению ферроптоза. Эффект не являлся аддитивным (см. рис. 5).
Ранее было показано, что устойчивость к ферроптозу провоспалительных макрофагов М1 RAW 264.7 обусловлена экспрессией индуцибельной iNOS и, соответственно, продукцией NO [20]. Оксид азота может быть добавлен к клеткам в виде доноров NO — веществ, которые высвобождают газообразный NO при попадании в среду с рН=7,4. Авторы использовали доноры NO c большим временем полураспада — DPTA (3 ч при 37оС) и Diethylenetriamine NONOate (20 ч при 37оС), тогда как время развития ферроптоза составляло 5 ч. Добавление доноров NO к мышиным макрофагам увеличивало их устойчивость к ферроптозу. Поскольку макрофаги человека продуцируют мало NO [28], мы поставили задачу изучить, как экзогенные доноры NO влияют на ферроптоз ТНР-1-макрофагов.
Мы сравнили эффекты доноров с разными временами полураспада при 37оС — с малым временем полураспада DEA NONOate (2 мин) и с относительно большим временем полураспада DPTA NONOate (3 ч). Доноры добавляли однократно после индуктора в концентрации 100 мкМ или несколько раз до достижения заданной концентрации. DEA NONOate не влиял на ферроптоз ТНР-1-макрофагов, тогда как DPTA полностью ингибировал ферроптоз как в случае однократной добавки, так и при добавлении 50 мкМ два раза. Быстрый распад DEA NONOate приводит к тому, что продуцируемый NO, видимо, не успевает попасть в клетки. Ферроптоз ТНР-1-макрофагов начинается только через ~5 ч после добавления индуктора. Тот факт, что однократная добавка DPTA (100 мкМ) в начале эксперимента полностью ингибировала ферроптоз, позволяет предположить, что механизм действия NO может быть следующим: оксид азота либо модифицирует белки-участники ферроптоза (например, взаимодействует с железом в активном центре липоксигеназы-15), либо входит в состав динитрозильных комплексов железа с последующим медленным высвобождением [29].
Заключение
Тремя независимыми методами анализа жизнеспособности клеток было показано, что ингибиторы GPX4 дозозависимо индуцируют клеточную гибель ТНР-1-макрофагов. Ингибирование смерти специфическим ингибитором Fer-1, а также детекция окисления липидов мембран клеток флуоресцентным маркером BODIPY C11 доказывают, что клетки погибают путем программируемой ферроптотической формы смерти. Одновременная обработка клеток индукторами ферроптоза с разным механизмом действия усиливает ферроптоз. В результате проведенного исследования показано, что NO, продуцируемый DPTA NONOate с периодом полураспада 3 ч, ингибирует ферроптоз в макрофагах, который развивается около 17–19 ч. Следовательно, существуют механизмы пролонгирования действия NO в клетках. Изучение механизмов ингибирования ферроптоза донорами NO имеет важное значение для дальнейшего использования этих соединений с целью регуляции ферроптоз-зависимых патологий.
Финансирование. Работа выполнена при финансовой поддержке гранта Российского научного фонда №23-25-00497 (https://rscf.ru/project/23-25-00497/).
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Литература
- Dixon S.J., Olzmann J.A. The cell biology of ferroptosis. Nat Rev Mol Cell Biol 2024; 25(6): 424–442, https://doi.org/10.1038/s41580-024-00703-5.
- Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascón S., Hatzios S.K., Kagan V.E., Noel K., Jiang X., Linkermann A., Murphy M.E., Overholtzer M., Oyagi A., Pagnussat G.C., Park J., Ran Q., Rosenfeld C.S., Salnikow K., Tang D., Torti F.M., Torti S.V., Toyokuni S., Woerpel K.A., Zhang D.D. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017; 171(2): 273–285, https://doi.org/10.1016/j.cell.2017.09.021.
- Yang W.S., Stockwell B.R. Ferroptosis: death by lipid peroxidation. Trends Cell Biol 2016; 26(3): 165–176, https://doi.org/10.1016/j.tcb.2015.10.014.
- Kagan V.E., Tyurina Y.Y., Vlasova I.I., Kapralov A.A., Amoscato A.A., Anthonymuthu T.S., Tyurin V.A., Shrivastava I.H., Cinemre F.B., Lamade A., Epperly M.W., Greenberger J.S., Beezhold D.H., Mallampalli R.K., Srivastava A.K., Bayir H., Shvedova A.A. Redox epiphospholipidome in programmed cell death signaling: catalytic mechanisms and regulation. Front Endocrinol (Lausanne) 2021; 11: 628079, https://doi.org/10.3389/fendo.2020.628079.
- Stockwell B.R., Jiang X., Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol 2020; 30(6): 478–490, https://doi.org/10.1016/j.tcb.2020.02.009.
- Jiang X., Stockwell B.R., Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021; 22(4): 266–282, https://doi.org/10.1038/s41580-020-00324-8.
- Bayır H., Dixon S.J., Tyurina Y.Y., Kellum J.A., Kagan V.E. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol 2023; 19(5): 315–336, https://doi.org/10.1038/s41581-023-00689-x.
- Toppo S., Flohé L., Ursini F., Vanin S., Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta 2009; 1790(11): 1486–1500, https://doi.org/10.1016/j.bbagen.2009.04.007.
- Yang Y., Wang Y., Guo L., Gao W., Tang T.L., Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis 2022; 13(4): 355, https://doi.org/10.1038/s41419-022-04775-z.
- Klyucherev T.O., Peshkova M.A., Revokatova D.P., Serejnikova N.B., Fayzullina N.M., Fayzullin A.L., Ershov B.P., Khristidis Y.I., Vlasova I.I., Kosheleva N.V., Svistunov A.A., Timashev P.S. The therapeutic potential of exosomes vs. matrix-bound nanovesicles from human umbilical cord mesenchymal stromal cells in osteoarthritis treatment. Int J Mol Sci 2024; 25(21): 11564, https://doi.org/10.3390/ijms252111564.
- Suleimanov S.K., Efremov Y.M., Klyucherev T.O., Salimov E.L., Ragimov A.A., Timashev P.S., Vlasova I.I. Radical-generating activity, phagocytosis, and mechanical properties of four phenotypes of human macrophages. Int J Mol Sci 2024; 25(3): 1860, https://doi.org/10.3390/ijms25031860.
- Daigneault M., Preston J.A., Marriott H.M., Whyte M.K., Dockrell D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 2010; 5(1): e8668, https://doi.org/ 10.1371/journal.pone.0008668.
- Balon K., Wiatrak B. PC12 and THP-1 cell lines as neuronal and microglia model in neurobiological research. Appl Sci 2021; 11(9): 3729, https://doi.org/10.3390/app11093729.
- Liang Y., Xu X.D., Xu X., Cai Y.B., Zhu Z.X., Zhu L., Ren K. Linc00657 promoted pyroptosis in THP-1-derived macrophages and exacerbated atherosclerosis via the miR-106b-5p/TXNIP/NLRP3 axis. Int J Biol Macromol 2023; 253(Pt 4): 126953, https://doi.org/10.1016/j.ijbiomac.2023.126953.
- Rossi Á.D., Higa L.M., Herlinger A.L., Ribeiro-Alves M., de Menezes M.T., Giannini A.L.M., Cardoso C.C., Da Poian A.T., Tanuri A., Aguiar R.S. Differential expression of human microRNAs during dengue virus infection in THP-1 monocytes. Front Cell Infect Microbiol 2021; 11: 714088, https://doi.org/10.3389/fcimb.2021.714088.
- Yi Z.H., Li S.Q., Ke J.Y., Wang Y., Zhao M.Z., Li J., Li M.Q., Zhu Z.L. Baicalein relieves ferroptosis-mediated phagocytosis inhibition of macrophages in ovarian endometriosis. Curr Issues Mol Biol 2022; 44(12): 6189–6204, https://doi.org/10.3390/cimb44120422.
- Hadian K. Ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 cooperatively suppress ferroptosis. Biochemistry 2020; 59(5): 637–638, https://doi.org/10.1021/acs.biochem.0c00030.
- Sun X., Ou Z., Chen R., Niu X., Chen D., Kang R., Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016; 63(1): 173–184, https://doi.org/10.1002/hep.28251.
- Shah R., Margison K., Pratt D.A. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem Biol 2017; 12(10): 2538–2545, https://doi.org/10.1021/acschembio.7b00730.
- Kapralov A.A., Yang Q., Dar H.H., Tyurina Y.Y., Anthonymuthu T.S., Kim R., St Croix C.M., Mikulska-Ruminska K., Liu B., Shrivastava I.H., Tyurin V.A., Ting H.C., Wu Y.L., Gao Y., Shurin G.V., Artyukhova M.A., Ponomareva L.A., Timashev P.S., Domingues R.M., Stoyanovsky D.A., Greenberger J.S., Mallampalli R.K., Bahar I., Gabrilovich D.I., Bayır H., Kagan V.E. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 2020; 16(3): 278–290, https://doi.org/10.1038/s41589-019-0462-8.
- Dar H.H., Anthonymuthu T.S., Ponomareva L.A., Souryavong A.B., Shurin G.V., Kapralov A.O., Tyurin V.A., Lee J.S., Mallampalli R.K., Wenzel S.E., Bayir H., Kagan V.E. A new thiol-independent mechanism of epithelial host defense against Pseudomonas aeruginosa: iNOS/NO• sabotage of theft-ferroptosis. Redox Biol 2021; 45: 102045, https://doi.org/10.1016/j.redox.2021.102045.
- Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci 2004; 75(6): 639–653, https://doi.org/10.1016/j.lfs.2003.10.042.
- Korhonen R., Lahti A., Kankaanranta H., Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy 2005; 4(4): 471–479, https://doi.org/10.2174/1568010054526359.
- Schairer D.O., Chouake J.S., Nosanchuk J.D., Friedman A.J. The potential of nitric oxide releasing therapies as antimicrobial agents. Virulence 2012; 3(3): 271–279, https://doi.org/10.4161/viru.20328.
- Martin-Sanchez D., Ruiz-Andres O., Poveda J., Carrasco S., Cannata-Ortiz P., Sanchez-Niño M.D., Ruiz Ortega M., Egido J., Linkermann A., Ortiz A., Sanz A.B. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol 2017; 28(1): 218–229, https://doi.org/10.1681/ASN.2015121376.
- Zhang Y., Sun C., Zhao C., Hao J., Zhang Y., Fan B., Li B., Duan H., Liu C., Kong X., Wu P., Yao X., Feng S. Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res 2019; 1706: 48–57, https://doi.org/10.1016/j.brainres.2018.10.023.
- Hoppenbrouwers T., Bastiaan-Net S., Garssen J., Pellegrini N., Willemsen L.E.M., Wichers H.J. Functional differences between primary monocyte-derived and THP-1 macrophages and their response to LCPUFAs. PharmaNutrition 2022; 22: 100322, https://doi.org/10.1016/j.phanu.2022.100322.
- Schneemann M., Schoeden G. Macrophage biology and immunology: man is not a mouse. J Leukoc Biol 2007; 81(3): 579; discussion 580, https://doi.org/10.1189/jlb.1106702. Erratum in: J Leukoc Biol 2007; 81(5): 1334, https://doi.org/10.1189/jlb.81.5.1334.
- Shumaev K.B., Dudylina A.L., Ivanova M.V., Pugachenko I.S., Ruuge E.K. Dinitrosyl iron complexes: formation and antiradical action in heart mitochondria. Biofactors 2018; 44(3): 237–244, https://doi.org/10.1002/biof.1418.








