Splicing-Sensitive DNA-Microarrays: Peculiarities and Applicationin Biomedical Research (Review)
Alternative splicing (АS) provides a variety of protein and mature mRNA isoforms encoded by a single gene, and is the essential component of cell and tissue differentiation and functioning. DNA-microarrays are highly productive transcriptome research technique both at the level of total gene expression assessment and alternatively spliced mRNA isoforms exploration. The study of AS patterns requires thorough probe design to achieve appropriate accuracy of the analysis.
There are two types of splicing-sensitive DNA-microarrays. The first type contain probes targeted to internal exonic sequences (exon bodies); the second type contain probes targeted to exon bodies and exon–exon and exon–intron junctions. So, the first section focused on probe sequence design, general features of splicing-sensitive DNA-microarrays and their main advantages and limitations.
The results of AS research obtained using DNA-microarrays have been reviewed in special section. In particular, DNA-microarrays were used to reveal a number pre-mRNA processing and splicing mechanisms, to investigate AS patterns associated with cancer, cell and tissue differentiation. Splicing machinery regulation was demonstrated to be an essential step during carcinogenesis and differentiation. The examples of application of splicing-sensitive DNA-microarrays for diagnostic markers discovering and pathology mechanism elucidation were also reviewed. Investigations of AS role in pluripotency, stem cell commitment, immune and infected cells functioning during immune response are the promising future directions. Splicing-sensitive DNA-microarrays are relatively inexpensive but powerful research tool that give reason to suppose their introduction in clinical practice within the next few years.
More than 90% human genes’ pre-messenger RNA (pre-mRNA) undergo alternative splicing (AS) [1, 2]. Translation products encoded by single gene mRNA isoforms resulted from AS are involved in different cell functions up to antagonistic. AS is among basic mechanisms involved in cell and tissue functioning and differentiation. There are 8 AS types (Figure 1). The main AS result is the variety of combinations of constitutive and alternative exons composing a mature transcript. Constitutive exons usually are present in all mature mRNA isoforms, while the presence of alternative exons is determined by the composition and functioning of splicing machinery. Thus, AS results in mRNA diversity forming a cell transcriptome.
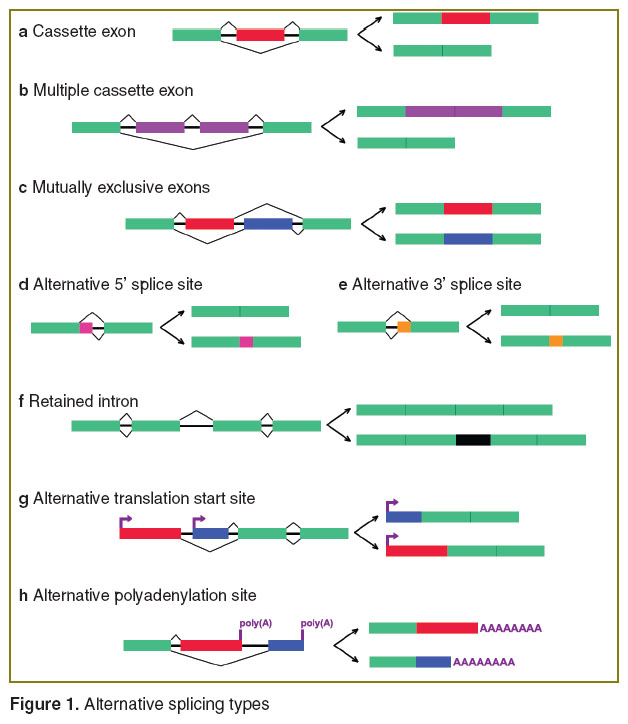 Figure 1. Alternative splicing types Figure 1. Alternative splicing types
|
Transcriptome research is conducted in two directions: 1) assessment of the total mRNA level of a gene (gene expression); 2) analysis of composition and quantitative distribution of mature mRNA spliced isoforms. Complex transcriptome investigations require high-throughput and accurate techniques. Real-time RT-PCR and PCR are time- and labor-consuming and enable to analyze only few genes within single experiment. However, they are considered as the “gold standard” of accuracy. Currently, there is a growing use of relatively new high efficient methods to study cell and tissue transcriptome: DNA-microarrays and RNA-sequencing (the latter is based on pyrosequencing technology) [3, 4]. There are no considerable differences in the accuracy of these two techniques, however, RNA-sequencing is more expensive [5–7]. Thus, DNA-microarrays provide an optimal combination of productivity, costs and accuracy of obtained results.
The present paper considers the current approaches to design of splicing-sensitive DNA-microarrays and reviews the key directions of their application in biological and medical studies.
Design features of splicing-sensitive DNA-microarrays
Selection of probes complementary to individual mRNA isoform sequences is the key step in design of splicing-sensitive DNA-microarrays. To achieve the highest analysis accuracy probes should meet the following requirements: 1) the narrow range of melting temperature (Tm) and nucleotide G and C percentage (%GC); 2) minimal possibility of a stable secondary structure (hairpin) formation; 3) minimal possibility of cross-hybridization (unspecific mRNA binding).
The important aspect of AS examination is the selection of probe hybridization areas within assessed mRNA. There are two types of probe binding localization: type 1 — inside exons and retained introns; type 2 — in exon–exon or exon–intron junctions (Figure 2). Type 1 probes have the higher possibility of successful selection of probes meeting aforementioned criteria due to relatively long sequences of human exons and introns [8]. Type 2 probes enable to make unambiguous conclusions regarding distribution and abundance level of spliced mRNAs. However, due to localization restrictions for type 2, selection of probes satisfying the above mentioned criteria is more difficult. Thus, DNA-microarrays containing type 1 probes and DNA-microarrays with both type probes are applied in AS research.
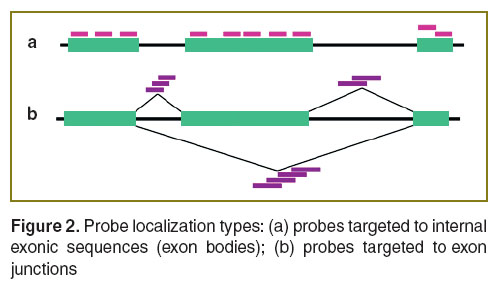 Figure 2. Probe localization types: (а) probes targeted to internal exonic sequences (exon bodies); (b) probes targeted to exon junctions Figure 2. Probe localization types: (а) probes targeted to internal exonic sequences (exon bodies); (b) probes targeted to exon junctions
|
Exon-targeted microarrays. The type 1 probe design is implemented in Exon 1.0 ST Array (Affymetrix, USA) the most widely used commercial splicing-sensitive microarray to date. The microarray contains over 5.5 million probes to detect and quantify the vast majority of annotated exons. Each exon corresponds to a set of 4 probes, length of each probe is 25 bases [9]. Despite high density of transcriptome coverage the microarray has a number of disadvantages: 1) relatively short probe length enhances cross hybridization probability; 2) high heterogeneity of probes (nucleotide G and C percentage varies from 0 to 100) results in a wide range of signal intensity, and complicates data analysis [10]; 3) inability to detect relatively small (less than 25 bases) 5’- and 3’-splicing sites shifts (See Figure 1 (d) and (e)).
The major problem stems from these “congenital” defects of Affymetrix microarrays is a high percentage of false-positive results. The percentage of confirmed AS events (by RT-PCR, qPCR) lies within the range of 20–80% [11–13].
The splicing index (SI) — the exon expression assessment criterion proposed by Affymetrix is calculated as follows:


First, normalized intensity (NI) of exon signal is calculated. Averaged signal level of the probes corresponding to exon i (ei) is divided by total intensity of all exons of gene j (1). Splicing index is calculated as a logarithm from ratio of normalized intensities of test and control (2) [14]. Due to heterogeneity of probes, gene expression level variations, unknown and predicted spliced isoforms presence this criterion results in a considerable percentage of inaccurate conclusions. Therefore, some investigations using Exon 1.0 ST Array is aimed to improved algorithm development and strict criteria introduction for correct signal/background discrimination, increasing reproducibility and specificity [15, 16]. The development of new algorithms and software for data processing, analysis and signal/background correction in experiments using exon-targeted Affymetrix microarrays is a growing segment of bioinformatics [17–19].
Studies using Agilent Exon Arrays are presented much rarely. The main difference of Agilent products is probe length — 50–60 bases. The shortest analyzed exons are 35 bases in length, probes corresponding to such “short” exons are filled up to 50 bases using poly-Т-spacers. Thus, exome (pool of all exons) coverage in Agilent Exon Arrays is significantly lower. In Human Exon 1.0 ST Array (Affymetrix) the number of exons covered (≥25 nucleobases in length) is 1,084,639, while the number of exon probes in Agilent Human Exon 400K Microarray is 233,164 [9, 20]. There are just a few reports in literature concerning the accuracy of Agilent Exon Arrays. The study of transcriptome changes in human mesenchymal stem cells in response to severe hypoxia revealed 53 alternatively spliced genes, the authors chose only 2 genes for validation: the presence of cassette exons was successfully confirmed [21].
Exon junction-targeted DNA-microarrays. Microarrays, containing probes complementary to both exon bodies and exon junctions are more informative compared to the previous microarray type. First, the probes for hybridization with junction sites enable to clearly detect all AS events. Second, combined data on probe hybridization with constitutive and alternative exons allows drawing more reliable inferences about AS events. For example, exon inclusion/skipping degree can be detected not only from change in probe hybridization intensity with the corresponding exon, but also from hybridization signal alterations of adjacent constitutive and alternative junctions (Figure 3) that is an additional selection criterion for true positive results. So, the study of AS of 16 clinically relevant tumor-associated genes showed 100% true positive results using combined data from all AS-targeted probes [22]. Selection of coordinated alterations in hybridization intensity of probes discriminating specific AS events provides ~75% analysis accuracy [23, 24].
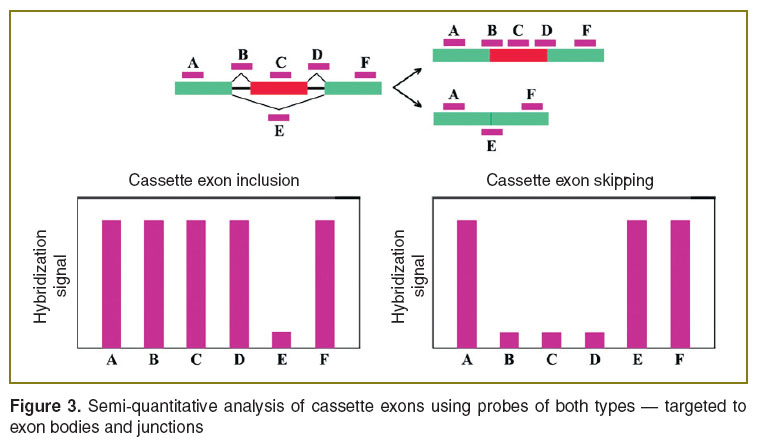 Figure 3. Semi-quantitative analysis of cassette exons using probes of both types — targeted to exon bodies and junctions Figure 3. Semi-quantitative analysis of cassette exons using probes of both types — targeted to exon bodies and junctions
|
Despite the clear advantages of the junction-targeted probes their design is substantially difficult. Localization of probes on a border sites severely limits the number of freedom degrees to select the sequences with a narrow range of Tm and %GC. Localization on a border sites also imposes limitations on probe length. Too long probe will hybridize effectively by one of its “halves” with a target exon in all spliced isoforms, containing this exon (Figure 4). On the contrary, a short-length probe will hybridize less effectively resulting in decrease of the analysis sensitivity. Srinivasan et al. [25] found that 36–40 bases is an optimal length for probes targeted to the exon–exon or exon–intron junctions. On the other hand, pilot trials of hGWSA (human Genome Wide Splicing Array; ExonHit, USA–France) showed that satisfactory specificity is provided by relatively short probes, 24–25 bases [26]. Primarily, these differences can be due to the type of hybridized nucleic acid. So, in the first example, DNA was hybridized on microarrays, while RNA was hybridized on hGWSA. The experimental material suggests that DNA provides higher sensitivity and specificity compared to RNA [27]. Splicing-sensitive Affymetrix microarrays contain 25-nucleotide probes hybridized with a DNA at temperature 45°С [28]. Agilent microarrays contain 60-nucleotide probes and are hybridized at 60–65°С [29–33]. Junction-targeted probe design strategy different from “search the best among possible” is uniform coverage of the junction site. In an experimental array based on Agilent platform, each junction area was uniformly covered by five 36-base probes with a step of 9 bases, validation showed 70–85% accuracy [34]. Human Transcriptome Array 2.0 (Affymetrix) has probes for 260,488 junctions; each junction is covered by four probes with a spacing of 2 bases [35].
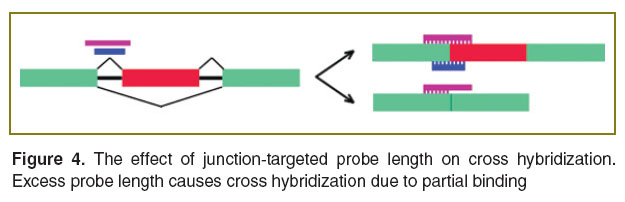 Figure 4. The effect of junction-targeted probe length on cross hybridization. Excess probe length causes cross hybridization due to partial binding Figure 4. The effect of junction-targeted probe length on cross hybridization. Excess probe length causes cross hybridization due to partial binding
|
Thus, despite the difficulties resulted from thermodynamic limitations, the probes targeted on exon–exon or exon–intron junctions provide high accuracy of AS event estimation compared to the exon-oriented probes. Therefore, vast majority of experimental splicing-sensitive microarrays have probes of both types.
AS research results obtained with the application of DNA-microarrays
Pre-mRNA processing and splicing mechanisms. Pre-mRNA processing mechanisms exploration and the revealing of target genes of splicing factors appeared among the first fundamental problems investigated with using splicing-sensitive DNA-microarrays. The study by J.C. Castle et al. [30] involving 48 human tissues and cell lines found over 9,500 AS events. Quantification of expression levels of splicing factors and cassette exons, combined with pre-mRNA sequences analysis revealed or confirmed the presence of 143 splicing cis-elements.
This direction has been further developed using an experimental splicing-sensitive Agilent microarray containing the probes to assess 1,804 AS events in 482 genes associated with pre-mRNA processing and oncogenesis [31]. In particular, target genes of CPEB1 (regulator responsible for the selection of alternative sites of pre-mRNA polyadenylation) have been discovered, splicing-associated and splicing-independent mechanisms of polyadenylation site displacement were also confirmed [33]. In the study of the antiproliferative mechanism of spliceostatin A (SSA) the model of pre-mRNA interaction with SF-3b-155 and U2 snRNP spliceosome elements has been proposed, the interfering influence of SSA molecule on this interaction has been also described [36]. In the study of Solier et al. [37, 38] camptothecin (CPT, topoisomerase I inhibitor) treatment of HCT116 cells caused the changes of splicing pattern of 998 genes, 140 of which participated in pre-mRNA splicing and processing regulation. There has been suggested the following mechanism: topoisomerase I inhibition results in RNA-polymerase II hyperphosphorylation, the decrease of elongation rate and the malfunction of splicing factors associated with RNA-polymerase II. Dutertre et al. [39] in their work showed that topoisomerase inhibitors cause AS of 3’-exons due to interference of alternative 3’-exon binding to HuR/ELAVL1 spliceosome component.
AS mechanisms involving RNA-helicases Ddx5 и Ddx17 which are also the spliceosome components [40], were studied using Human Exon Array [41]. Ddx5/17 cooperation with nuclear ribonucleoprotein hnRNP H/F was found to provide its interaction with “strong” (primarily recognized) splicing sites and inclusion of the exons flanked by strong splicing sites in a mature mRNA. Moreover, there has been shown the role of Ddx5/17 as a splicing regulator during intracellular signaling in response to the binding of steroid hormones to their receptors [41, 42].
Large-scale investigations using microarrays targeted to exon junctions enabled to determine basic functioning mechanisms of primary splicing factors: SRSF1 and SRSF2 [43], PTB [44], hnRNP family [45]. These regulators mediate exon inclusion or skipping that depends on many factors: strength and localization of splicing sites, the length of adjacent introns and spliceosome composition.
Tumor-associated AS patterns. Currently, investigations focused on total differential gene expression during carcinogenesis make up the most extensive area of DNA-microarray application. However, works devoted to the study of tumor-associated splicing patterns are rather rare. Detection of specific spliced isoforms typical for a certain tumor type is a robust approach to the finding of the new therapeutic and prognostic markers and target drug development.
The studies of lung cancer [46, 47], colon cancer [48], brain cancers [49, 50], leukemia [51–54], Hodgkin’s lymphoma [55], head and neck squamous cell carcinoma [56] showed that genes of cytoskeleton elements, cell cycle realization and control, cell-cell interaction, apoptosis-associated genes are most frequently undergo AS during carcinogenesis.
The number of AS researches carried out using DNA-microarrays focused on the tumor-associated mechanisms of pre-mRNA splicing and processing. A large-scale study by Sveen et al. [57, 58] comprising 7 solid tumor types considers transcriptome instability to be the general characteristic of carcinogenesis due to the decreased expression of splicing factors. Similar results were obtained by Carrigan et al. [59]. The decline of spliced isofoms variety as well as the decrease of expression level of 28 spliceosomal genes in pancreatic carcinoma cell lines Capan-1 and MiaPaCa2 compared to pancreatic duct cells HPDE6 were observed in this study. Excess activation of transcription factor c-Jun is associated with oncogenic transformation and causes resistance to apoptosis [60–63]. Knockout of c-Jun in mice breast epithelium cells resulted in expression changes of 114 genes involved in pre-mRNA processing and splicing. In addition, many apoptosis associated genes (Сasp-8, Casp-9, Kifap3, Wisp1, etc.) were observed to undergo AS [64]. The authors proposed the mechanism of oncogenic impact of c-Jun inhibiting a pro-apoptotic splicing factor SRSF2 and attenuating spliceosome efficiency. The study of 31 breast tumor cell lines belonging to 3 subtypes revealed similar AS patterns in each subtype [65]. Tissue specific splicing factor Fox2 was shown to be one of the master regulators determining AS patterns specific for each subtype.
AS patterns associated with cell and tissue differentiation. Cell and tissue differentiation is executed through gene expression regulation both on a quantitative level and also by changing AS patterns resulting in expression of tissue-specific protein isoforms. Tissue-specific splicing is accomplished by coordinated activity of general splicing factors (SR, hnRNP protein families) and specific factors, such as Nova, nPTB, Fox, nSR100, MBNL, CELF [66–68]. Warzecha et al. [69] identified splicing factors ESRP1 and ESRP2 responsible for epithelial tissue differentiation and the maintenance of epithelial phenotype. The authors [70, 71] determined the spectrum of ESRP1 and ESRP2 target genes, the key motif of cis-elements — UGG, the principle of splicing regulation: cis-elements in downstream intron lead to exon inclusion, while being within exon, they lead to its skipping. ESRP1 and ESRP2 knockout results in dramatic alterations of splicing patterns and the epithelial phenotype loss that was interpreted by the authors as the epithelial-mesenchymal transition. Similarly, there were analyzed the peculiarities of splicing regulated by Fox factor specific to the brain, heart and striated muscles. The authors studied the spectrum of Fox target genes, determined the main regulatory cis-element UGCAUG, which is an enhancer or silencer within downstream or upstream intron, respectively [72, 73].
Differentiation features and AS patterns in nervous tissue were also explored using splicing-sensitive microarrays. The differentiation of murine embryonic carcinoma cells Р19 to neurons induced by trans-retinoic acid was accompanied by the appearance of neurospecific splicing factor nPTB and the disappearance of a general splicing repressor PTB. Splicing of 26% exons (54 of 202) during differentiation was found to be regulated by a neurospecific factor nPTB [74]. A large comparative study of brain transcriptome in intact mice and Nova (brain-specific splicing regulator) knockout mice revealed about 600 AS events. These findings combined with the cis-regulatory element analysis allowed to draw the following conclusions: а) Nova cis-elements localized within downstream intron are enhancers, whereas localized within exon and upstream intron are splicing silencers; b) Nova and Fox factors co-regulate about 100 exons; c) Nova executes two-level regulation of protein-protein interactions involved in synaptic contacts: splicing regulation of kinases and phosphatases (25 and 9, respectively) and also splicing regulation of the exons encoding phosphorylated protein sites [75].
Transcriptome analysis of three cell types — epitheliocytes, fibroblasts and endothelial cells — revealed cell type-specific AS patterns persisting independently of the tissue of origin. For example, epitheliocytes of the lungs, prostate, kidney, breast, and other organs had a common pattern of spliced isoforms. AS patterns characteristic for each cell type were based on the expression of tissue-specific splicing regulators. So, ESRP and hnRNP factors were specifically expressed in epitheliocytes, NOVA1 and RBFox2 were expressed in fibroblasts, PTBP and MBNL — in endothelial cells [76].
Thus, the research results clearly show that AS is a necessary tool to maintain cell and tissue identity.
Diagnostic markers discovering and pathology mechanism elucidation using splicing-sensitive microarrays. Since the greatest diversity of spliced isoforms is presented in central nervous system [14, 30], splicing-sensitive DNA-microarrays are used in researches and diagnostics of neurodegenerative diseases. For example, the search for mRNA splice variants in blood for early detection of Alzheimer’s and Parkinson’s diseases prior to clinical manifestations is conducted [77–79].
The comparison of age- and pathology-related changes in temporal lobe transcriptome [80] showed that Alzheimer’s disease and frontotemporal lobar degeneration development is associated with decreased Nova (splicing regulator) expression level. The decreased activity of the regulator was demonstrated to cause AS imbalance in 20 exons of neurospecific genes responsible for synaptic transmission. Neurodegenerative disease amyotrophic lateral sclerosis (ALS) results from mutation of TARDBP gene encoding TBP-43 spliceosome component [81]. The comparison of motor neuron transcriptome in healthy volunteers and ALS patients revealed the differences in splicing patterns of over 4,000 genes, most of which involved in cytoskeleton formation and cell shape maintenance [82].
The study of splicing patterns in distal muscles bioptates in patients with myotonic dystrophy enabled to confirm and discover the early disease markers. Aberrant enhanced binding of splicing regulator MBNL1 to amplified CUG- or CCUG-repeats within pre-mRNA of mutant genes DMPK or ZNF9/CNBP causes MBNL1 sequestration and splicing failure of the rest pre-mRNA targets of this regulator [83, 84]. Patients without marked clinical manifestations of the disease were found to have spliced isoforms of genes INSR, TTN, RYR1, CAMK2B, ARFGAP2 which are the targets of splicing regulator MBNL1 [85].
Thus, splicing-sensitive DNA-microarrays are the tools for new diagnostic markers search and pathogenesis investigation, but are not applied in clinical diagnostics. However, DNA-microarrays for genotyping are currently used as diagnostic tool. In 2010 the International Standard Cytogenomic Array Consortium recommended to use microarrays as a first-tier clinical diagnostic test for individuals with congenital anomalies instead of karyotyping [86]. In a number of countries DNA-microarrays are used in clinical diagnostic practice to reveal chromosome rearrangements associated with a wide spectrum of disorders [87–90]. In prospect, mRNA pools associated with various diseases and having diagnostic and prognostic value are expected to be validated, so the introduction of splicing-sensitive microarrays into clinical diagnostic practice is the matter of the near future.
Promising lines of research
Induced pluripotent stem cells (IPSC) obtaining by means of genetic engineering [91–93] has broadened the prospects for regenerative medicine. Currently, most researches concerning SC transcriptome are focused on the dynamics of total gene expression during SC differentiation and pluripotent state maintenance [94–98]. A few works concerning AS in IPSC indicate that the process is critical for SC pluripotent state maintenance [99, 100] and differentiation [101–103]. The study of AS processes in the course of IPSC induction and commitment using splicing-sensitive DNA-microarrays is expected to become a developing segment of biomedical research.
Another significant research segment remaining under investigated in AS context is an infection process modeling: the study of AS patterns in immunocytes and infected cells during infection process development and immune response. The dynamic transcriptome characteristics during differentiation and activation of various immunocytes subpopulations have been studied in detail [104–110]. The total gene expression changes have been analyzed in nervous system cells infected by Argentinean hemorrhagic fever virus [111], HSV-1 [112], in hepatocytes infected by HCV [113], in astrocytes during modeling bacterial infection [114], in epithelial enterocytes [115]. However, the AS regulation of key genes of immune response during infection process is poorly studied [116, 117] and requires further investigations.
Despite the growing use of RNA-sequencing technology, DNA-microarrays remain essential transcriptome research tool. Owing to the compromise between costs, value and amount of obtained data a marked increase of DNA-microarrays usage in the AS research as well as the introduction of splicing-sensitive microarrays in clinical practice are expected in the next decade.
Study Funding and Conflicts of Interest. The study was not funded by any sources, and the authors have no conflicts of interest related to the present study.
References
- Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456(7221): 470–476, http://dx.doi.org/10.1038/nature07509.
- Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high throughput sequencing. Nat Genet 2008; 40(12): 1413–1435, http://dx.doi.org/10.1038/ng.259.
- Novais R.C., Thorstenson Y.R. The evolution of Pyrosequencing® for microbiology: from genes to genomes. J Microbiol Methods 2011; 86(1): 1–7, http://dx.doi.org/10.1016/j.mimet.2011.04.006.
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10(1): 57–63, http://dx.doi.org/10.1038/nrg2484.
- Ramani A.K., Calarco J.A., Pan Q., Mavandadi S., Wang Y., Nelson A.C., Lee L.J., Morris Q., Blencowe B.J., Zhen M., Fraser A.G. Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res 2011; 21(2): 342–348, http://dx.doi.org/10.1101/gr.114645.110.
- Malone J.H., Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biology 2011; 9: 34, http://dx.doi.org/10.1186/1741-7007-9-34.
- RNA-Seq misses what HTA delivers. URL: http://www.affymetrix.com/fa/media/hta_array_2_0_flyer.pdf.
- Affymetrix. GeneChip® Exon Array design. URL: http://media.affymetrix.com/support/technical/technotes/exon_array_design_technote.pdf.
- Affymetrix. GeneChip® Exon Array System for human, mouse, and rat. URL: http://www.affymetrix.com/support/technical/datasheets/exon_arraydesign_datasheet.pdf.
- Jaksik R., Marczyk M., Polanska J., Rzeszowska-Wolny J. Sources of high variance between probe signals in Affymetrix short oligonucleotide microarrays. Sensors (Basel) 2014; 14(1): 532–548; http://dx.doi.org/10.3390/s140100532.
- Hung L.H., Heiner M., Hui J., Schreiner S., Benes V., Bindereif A. Diverse roles of hnRNP L in mammalian mRNA processing: a combined microarray and RNAi analysis. RNA 2008; 14(2): 284–296, http://dx.doi.org/10.1261/rna.725208.
- Kurokawa K., Akaike Y., Masuda K., Kuwano Y., Nishida K., Yamagishi N., Kajita K., Tanahashi T., Rokutan K. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene 2014; 33(11): 1407–1417, http://dx.doi.org/10.1038/onc.2013.86.
- Yamashita Y., Matsuura T., Shinmi J., Amakusa Y., Masuda A., Ito M., Kinoshita M., Furuya H., Abe K., Ibi T., Sahashi K., Ohno K. Four parameters increase the sensitivity and specificity of the exon array analysis and disclose 25 novel aberrantly spliced exons in myotonic dystrophy. J Hum Genet 2012; 57(6): 368–374, http://dx.doi.org/10.1038/jhg.2012.37.
- Clark T.A., Schweitzer A.C., Chen T.X., Staples M.K., Lu G., Wang H., Williams A., Blume J.E. Discovery of tissue specific exons using comprehensive human exon microarrays. Genome Biol 2007; 8(4): R64, http://dx.doi.org/10.1186/gb-2007-8-4-r64.
- Xing Y., Stoilov P., Kapur K., Han A., Jiang H., Shen S., Black D.L., Wong W.H. MADS: a new and improved method for analysis of differential alternative splicing by exon-tiling microarrays. RNA 2008; 14(8): 1470–1479, http://dx.doi.org/10.1261/rna.1070208.
- Risueño A., Roson-Burgo B., Dolnik A., Hernandez-Rivas J.M., Bullinger L., De Las Rivas J. A robust estimation of exon expression to identify alternative spliced genes applied to human tissues and cancer samples. BMC Genomics 2014; 15: 879, http://dx.doi.org/10.1186/1471-2164-15-879.
- Chen P., Lepikhova T., Hu Y., Monni O., Hautaniemi S. Comprehensive exon array data processing method for quantitative analysis of alternative spliced variants. Nucleic Acids Research 2011; 39(18): e123, http://dx.doi.org/10.1093/nar/gkr513.
- Kapetis D., Clarelli F., Vitulli F., de Rosbo N.K., Beretta O., Foti M., Ricciardi-Castagnoli P., Zolezzi F. AMDA 2.13: a major update for automated cross-platform microarray data analysis. Biotechniques 2012; 53(1): 33–40.
- Liu X., Gao Z., Zhang L., Rattray M. puma 3.0: improved uncertainty propagation methods for gene and transcript expression analysis. BMC Bioinformatics 2013; 14: 39, http://dx.doi.org/10.1186/1471-2105-14-39.
- Agilent Technologies. Comprehensive coverage with the Agilent SurePrint G3 Exon Microarray system. URL: http://www.agilent.com/cs/library/brochures/5990-6928en_lo.pdf.
- Hu X., Wu R., Shehadeh L.A., Zhou Q., Jiang C., Huang X., Zhang L., Gao F., Liu X., Yu H., Webster K.A., Wang J. Severe hypoxia exerts parallel and cell-specific regulation of gene expression and alternative splicing in human mesenchymal stem cells. BMC Genomics 2014; 15: 303, http://dx.doi.org/10.1186/1471-2164-15-303.
- Pesson M., Eymin B., De La Grange P., Simon B., Corcos L. A dedicated microarray for in-depth analysis of pre-mRNA splicing events: application to the study of genes involved in the response to targeted anticancer therapies. Molecular Cancer 2014, 13: 9, http://dx.doi.org/10.1186/1476-4598-13-9.
- Li C., Kato M., Shiue L., Shively J.E., Ares M. Jr., Lin R.J. Cell type and culture condition-dependent alternative splicing in human breast cancer cells revealed by splicing-sensitive microarrays. Cancer Res 2006; 66(4): 1990–1999, http://dx.doi.org/10.1158/0008-5472.can-05-2593.
- Zhou W., Calciano M.A., Jordan H., Brenner M., Johnson S., Wu D., Lei L., Pallares D., Beurdeley P., Rouet F., Gill P.S., Bracco L., Soucaille C., Einstein R. High resolution analysis of the human transcriptome: detection of extensive alternative splicing independent of transcriptional activity. BMC Genet 2009; 10: 63, http://dx.doi.org/10.1186/1471-2156-10-63.
- Srinivasan K., Shiue L., Hayes J.D., Centers R., Fitzwater S., Loewen R., Edmondson L.R., Bryant J., Smith M., Rommelfanger C., Welch V., Clark T.A., Sugnet C.W., Howe K.J., Mandel-Gutfreund Y., Ares M. Jr. Detection and measurement of alternative splicing using splicing-sensitive microarrays. Methods 2005; 37: 345–359, http://dx.doi.org/10.1016/j.ymeth.2005.09.007.
- Fehlbaum P., Guihal C., Bracco L., Cochet O. A microarray configuration to quantify expression levels and relative abundance of splice variants. Nucleic Acids Res 2005; 33(5): e47, http://dx.doi.org/10.1093/nar/gni047.
- Harrison A., Binder H., Buhot A., Burden C.J., Carlon E., Gibas C., Gamble L.J., Halperin A., Hooyberghs J., Kreil D.P., Levicky R., Noble P.A., Ott A., Pettitt B.M., Tautz D., Pozhitkov A.E. Physico-chemical foundations underpinning microarray and next-generation sequencing experiments. Nucleic Acids Res 2013; 41(5): 2779–2796, http://dx.doi.org/10.1093/nar/gks1358.
- Affymetrix. GeneChip® WT PLUS Reagent Kit. URL: http://media.affymetrix.com/support/downloads/manuals/wtplus_reagentkit_assay_manual.pdf.
- Agilent 2015. Agilent One-Color Microarray-Based Exon Analysis — Low Input Quick Amp WT Labeling. URL: http://www.agilent.com/cs/library/usermanuals/Public/G4140-90042_Exon_One-color_2.0.pdf.
- Castle J.C., Zhang C., Shah J.K., Kulkarni A.V., Kalsotra A., Cooper T.A., Johnson J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat Gen 2008; 40(12): 1416–1425, http://dx.doi.org/10.1038/ng.264.
- Muñoz M.J., Pérez Santangelo M.S., Paronetto M.P., de la Mata M., Pelisch F., Boireau S., Glover-Cutter K., Ben-Dov C., Blaustein M., Lozano J.J., Bird G., Bentley D., Bertrand E., Kornblihtt A.R. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 2009; 137(4): 708–720, http://dx.doi.org/10.1016/j.cell.2009.03.010.
- Paronetto M.P., Miñana B., Valcarcel J. The Ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol Cell 2011; 43(3): 353–368, http://dx.doi.org/10.1016/j.molcel.2011.05.035.
- Bava F.A., Eliscovich C., Ferreira P.G., Miсana B., Ben-Dov C., Guigó R., Valcárcel J., Méndez R. CPEB1 coordinates alternative 3’-UTR formation with translational regulation. Nature 2013; 495(7429): 121–125, http://dx.doi.org/10.1038/nature11901.
- Le K., Mitsouras K., Roy M., Wang Q., Xu Q., Nelson S.F., Lee C. Detecting tissue-specific regulation of alternative splicing as a qualitative change in microarray data. Nucleic Acids Res 2004; 32(22): e180, http://dx.doi.org/10.1093/nar/gnh173.
- Xu W., Seok J., Mindrinos M.N., Schweitzer A.C., Jiang H., Wilhelmy J., Clark T.A., Kapur K., Xing Y., Faham M., Storey J.D., Moldawer L.L., Maier R.V., Tompkins R.G., Wong W.H., Davis R.W., Xiao W.; Inflammation and Host Response to Injury Large-Scale Collaborative Research Program. Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci USA 2011; 108(9): 3707–3712, http://dx.doi.org/10.1073/pnas.1019753108.
- Corrionero A., Miñana B., Valcárcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Gen Dev 2011; 25(5): 445–459, http://dx.doi.org/10.1101/gad.2014311.
- Solier S., Barb J., Zeeberg B.R., Varma S., Ryan M.C., Kohn K.W., Weinstein J.N., Munson P.J., Pommier Y. Genome-wide analysis of novel splice variants induced by topoisomerase I poisoning shows preferential occurrence in genes encoding splicing factors. Cancer Res 2010; 70(20): 8055–8065, http://dx.doi.org/10.1158/0008-5472.CAN-10-2491.
- Solier S., Ryan M.C., Martin S.E., Varma S., Kohn K.W., Liu H., Zeeberg B.R., Pommier Y. Transcription poisoning by Topoisomerase I is controlled by gene length, splice sites, and miR-142-3p. Cancer Res 2013; 73(15): 4830–4839, http://dx.doi.org/10.1158/0008-5472.CAN-12-3504.
- Dutertre M., Chakrama F.Z., Combe E., Desmet F.O., Mortada H., Polay Espinoza M., Gratadou L., Auboeuf D. A recently evolved class of alternative 3’-terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nat Commun 2014; 5: 3395, http://dx.doi.org/10.1038/ncomms4395.
- Liu Z.-R. p68 RNA helicase is an essential human splicing factor that acts at the U1 snRNA-5’ splice site duplex. Mol Cell Biol 2002; 22(15): 5443–5450, http://dx.doi.org/10.1128/mcb.22.15.5443-5450.2002.
- Dardenne E., Polay Espinoza M., Fattet L., Germann S., Lambert M.P., Neil H., Zonta E., Mortada H., Gratadou L., Deygas M., Chakrama F.Z., Samaan S., Desmet F.O., Tranchevent L.C., Dutertre M., Rimokh R., Bourgeois C.F., Auboeuf D. RNA helicases DDX5 and DDX17 dynamically orchestrate transcription, miRNA, and splicing programs in cell differentiation. Cell Rep 2014; 7(6): 1900–1913, http://dx.doi.org/10.1016/j.celrep.2014.05.010.
- Samaan S., Tranchevent L.-C., Dardenne E., Espinoza M.P., Zonta E., Germann S., Gratadou L., Dutertre M., Auboeuf D. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic Acids Res 2014; 42(4): 2197–2207, http://dx.doi.org/10.1093/nar/gkt1216.
- Pandit S., Zhou Y., Shiue L., Coutinho-Mansfield G., Li H., Qiu J., Huang J., Yeo G.W., Ares M., Fu X.-D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell 2013; 50(2): 223–235, http://dx.doi.org/10.1016/j.molcel.2013.03.001.
- Huelga S.C., Vu A.Q., Arnold J.D., Liang T.Y., Liu P.P., Yan B.Y., Donohue J.P., Shiue L., Hoon S., Brenner S., Ares M. Jr., Yeo G.W. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 2012; 1(2): 167–178, http://dx.doi.org/10.1016/j.celrep.2012.02.001.
- Llorian M., Schwartz S., Clark T.A., Hollander D., Tan L.Y., Spellman R., Gordon A., Schweitzer A.C., de la Grange P., Ast G., Smith C.W. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol 2010; 17(9): 1114–1123, http://dx.doi.org/10.1038/nsmb.1881.
- Xi L., Feber A., Gupta V., Wu M., Bergemann A.D., Landreneau R.J., Litle V.R., Pennathur A., Luketich J.D., Godfrey T.E. Whole genome exon arrays identify differential expression of alternatively spliced, cancer-related genes in lung cancer. Nucleic Acids Res 2008; 36(20): 6535–6547, http://dx.doi.org/10.1093/nar/gkn697.
- de Miguel F.J., Sharma R.D., Pajares M.J., Montuenga L.M., Rubio A., Pio R. Identification of alternative splicing events regulated by the oncogenic factor SRSF1 in lung cancer. Cancer Res 2014; 74(4): 1105–1115, http://dx.doi.org/10.1158/0008-5472.CAN-13-1481.
- Gardina P.J., Clark T.A., Shimada B., Staples M.K., Yang Q., Veitch J., Schweitzer A., Awad T., Sugnet C., Dee S., Davies C., Williams A., Turpaz Y. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics 2006; 7(1): 325, http://dx.doi.org/10.1186/1471-2164-7-325.
- Guo X., Chen Q.R., Song Y.K., Wei J.S., Khan J. Exon array analysis reveals neuroblastoma tumors have distinct alternative splicing patterns according to stage and MYCN amplification status. BMC Med Genomics 2011; 4: 35, http://dx.doi.org/10.1186/1755-8794-4-35.
- Yu F., Fu W.-M. Identification of differential splicing genes in gliomas using exon expression profiling. Mol Med Rep 2015; 11(2): 843–850, http://dx.doi.org/10.3892/mmr.2014.2775.
- Gerber J.M., Gucwa J.L., Esopi D., Gurel M., Haffner M.C., Vala M., Nelson W.G., Jones R.J., Yegnasubramanian S. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget 2013; 4(5): 715–728, http://dx.doi.org/10.18632/oncotarget.990.
- Adamia S., Haibe-Kains B., Pilarski P.M., Bar-Natan M., Pevzner S., Avet-Loiseau H., Lode L., Verselis S., Fox E.A., Burke J., Galinsky I., Dagogo-Jack I., Wadleigh M., Steensma D.P., Motyckova G., Deangelo D.J., Quackenbush J., Stone R., Griffin J.D. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res 2014; 20(5): 1135–1145, http://dx.doi.org/10.1158/1078-0432.CCR-13-0956.
- Adamia S., Bar-Natan M., Haibe-Kains B., Pilarski P.M., Bach C., Pevzner S., Calimeri T., Avet-Loiseau H., Lode L., Verselis S., Fox E.A., Galinsky I., Mathews S., Dagogo-Jack I., Wadleigh M., Steensma D.P., Motyckova G., Deangelo D.J., Quackenbush J., Tenen D.G., Stone R.M., Griffin J.D. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): new potential targets in AML. Blood 2014; 123(18): 2816–2825, http://dx.doi.org/10.1182/blood-2013-02-481507.
- Relógio A., Ben-Dov C., Baum M., Ruggiu M., Gemund C., Benes V., Darnell R.B., Valcárcel J. Alternative splicing microarrays reveal functional expression of neuron-specific regulators in Hodgkin lymphoma cells. J Biol Chem 2005; 280(6): 4779–4784, http://dx.doi.org/10.1074/jbc.m411976200.
- Li R., Ochs M.F., Ahn S.M., Hennessey P., Tan M., Soudry E., Gaykalova D.A., Uemura M., Brait M., Shao C., Westra W., Bishop J., Fertig E.J., Califano J.A. Expression microarray analysis reveals alternative splicing of LAMA3 and DST genes in head and neck squamous cell carcinoma. PLoS One 2014; 9(3): e91263, http://dx.doi.org/10.1371/journal.pone.0091263.
- Liu J., Xiao Y., Xiong H.-M., Li J., Huang B., Zhang H.-B., Feng D.-Q., Chen X.-M., Wang X.-Z. Alternative splicing of apoptosis-related genes in imatinib-treated K562 cells identified by exon array analysis. Int J Mol Med 2012; 29(4): 690–698, http://dx.doi.org/10.3892/ijmm.2011.872.
- Sveen A., Еgesen T.H., Nesbakken A., Rognum T.O., Lothe R.A., Skotheim R.I. Transcriptome instability in colorectal cancer identified by exon microarray analyses: Associations with splicing factor expression levels and patient survival. Genome Med 2011; 3(5): 32, http://dx.doi.org/10.1186/gm248.
- Sveen A., Johannessen B., Teixeira M.R., Lothe R.A., Skotheim R.I. Transcriptome instability as a molecular pan-cancer characteristic of carcinomas. BMC Genomics 2014; 15: 672, http://dx.doi.org/10.1186/1471-2164-15-672.
- Carrigan P.E., Bingham J.L., Srinvasan S., Brentnall T.A., Miller L.J. Characterization of alternative spliceoforms and the RNA splicing machinery in pancreatic cancer. Pancreas 2011; 40(2): 281–288, http://dx.doi.org/10.1097/MPA.0b013e31820128d2.
- Katiyar S., Casimiro M.C., Dettin L., Ju X., Wagner E.F., Tanaka H., Pestell R.G. C-jun inhibits mammary apoptosis in vivo. Mol Biol Cell 2010; 21(23): 4264–4274, http://dx.doi.org/10.1091/mbc.E10-08-0705.
- Albanese C., Johnson J., Watanabe G., Eklund N., Vu D., Arnold A., Pestell R.G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem 1995; 270(40): 23589–23597, http://dx.doi.org/10.1074/jbc.270.40.23589.
- Eferl R., Ricci R., Kenner L., Zenz R., David J.P., Rath M., Wagner E.F. Liver tumor development: c-Jun antagonizes the proapoptotic activity of p53. Cell 2003; 112(2): 181–192, http://dx.doi.org/10.1016/s0092-8674(03)00042-4.
- Shaulian E., Schreiber M., Piu F., Beeche M., Wagner E.F., Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000; 103(6): 897–907, http://dx.doi.org/10.1016/s0092-8674(00)00193-8.
- Katiyar S., Jiao X., Addya S., Ertel A., Covarrubias Y., Rose V., Casimiro M.C., Zhou J., Lisanti M.P., Nasim T., Fortina P., Pestell R.G. Mammary gland selective excision of c-Jun identifies its role in mRNA splicing. Cancer Res 2012; 72(4): 1023–1034, http://dx.doi.org/10.1158/0008-5472.CAN-11-3647.
- Lapuk A., Marr H., Jakkula L., Pedro H., Bhattacharya S., Purdom E., Hu Z., Simpson K., Pachter L., Durinck S., Wang N., Parvin B., Fontenay G., Speed T., Garbe J., Stampfer M., Bayandorian H., Dorton S., Clark T.A., Schweitzer A., Wyrobek A., Feiler H., Spellman P., Conboy J., Gray J.W. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol Cancer Res 2010; 8(7): 961–974, http://dx.doi.org/10.1158/1541-7786.MCR-09-0528.
- Chen M., Manley J.L. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 2009; 10: 741–754, http://dx.doi.org/10.1038/nrm2777.
- Hartmann B., Valcárcel J. Decrypting the genome’s alternative messages. Curr Opin Cell Biol 2009; 21(3): 377–386, http://dx.doi.org/10.1016/j.ceb.2009.02.006.
- Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010; 463(7280): 457–463, http://dx.doi.org/10.1038/nature08909.
- Warzecha C.C., Sato T.K., Nabet B., Hogenesch J.B., Carstens R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 2009; 33(5): 591–601, http://dx.doi.org/10.1016/j.molcel.2009.01.025.
- Warzecha C.C., Shen S., Xing Y., Carstens R.P. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 2009; 6(5): 546–562, http://dx.doi.org/10.4161/rna.6.5.9606.
- Warzecha C.C., Jiang P., Amirikian K., Dittmar K.A., Lu H., Shen S., Guo W., Xing Y., Carstens R.P. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J 2010; 29(19): 3286–3300, http://dx.doi.org/10.1038/emboj.2010.195.
- Das D., Clark T.A., Schweitzer A., Yamamoto M., Marr H., Arribere J., Minovitsky S., Poliakov A., Dubchak I., Blume J.E., Conboy J.G. A correlation with exon expression approach to identify cis-regulatory elements for tissue-specific alternative splicing. Nucleic Acids Res 2007; 35(14): 4845–4857, http://dx.doi.org/10.1093/nar/gkm485.
- Zhang C., Zhang Z., Castle J., Sun S., Johnson J., Krainer A.R., Zhang M.Q. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 2008; 22(18): 2550–2563, http://dx.doi.org/10.1101/gad.1703108.
- Boutz P.L., Stoilov P., Li Q., Lin C.H., Chawla G., Ostrow K., Shiue L., Ares M. Jr., Black D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 2007; 21(13): 1636–1652, http://dx.doi.org/10.1101/gad.1558107.
- Zhang C., Frias M.A., Mele A., Ruggiu M., Eom T., Marney C.B., Wang H., Licatalosi D.D., Fak J.J., Darnell R.B. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 2010; 329(5990): 439–443, http://dx.doi.org/10.1126/science.1191150.
- Mallinjoud P., Villemin J.P., Mortada H., Polay Espinoza M., Desmet F.O., Samaan S., Chautard E., Tranchevent L.C., Auboeuf D. Endothelial, epithelial, and fibroblast cells exhibit specific splicing programs independently of their tissue of origin. Genome Res 2014; 24(3): 511–521, http://dx.doi.org/10.1101/gr.162933.113.
- Potashkin J.A., Santiago J.A., Ravina B.M., Watts A., Leontovich A.A. Biosignatures for Parkinson’s disease and atypical parkinsonian disorders patients. PLoS One 2012; 7(8): e43595, http://dx.doi.org/10.1371/journal.pone.0043595.
- Calciano M., Lemarié J.C., Blondiaux E., Einstein R., Fehlbaum-Beurdeley P. A predictive microarray-based biomarker for early detection of Alzheimer’s disease intended for clinical diagnostic application. Biomarkers 2013; 18(3): 264–272, http://dx.doi.org/10.3109/1354750X.2013.773083.
- Lai M.K.P., Esiri M.M., Tan M.G.K. Genome-wide profiling of alternative splicing in Alzheimer’s disease. Genomics Data 2014; 2: 290–292, http://dx.doi.org/10.1016/j.gdata.2014.09.002.
- Tollervey J.R., Wang Z., Hortobagyi T., Witten J.T., Zarnack K., Kayikci M., Clark T.A., Schweitzer A.C., Rot G., Curk T., Zupan B., Rogelj B., Shaw C.E., Ule J. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res 2011; 21(10): 1572–1582, http://dx.doi.org/10.1101/gr.122226.111.
- Pesiridis G.S., Lee V.M., Trojanowski J.Q. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Gen 2009; 18(R2): R156–R162, http://dx.doi.org/10.1093/hmg/ddp303.
- Highley J.R., Kirby J., Jansweijer J.A., Webb P.S., Hewamadduma C.A., Heath P.R., Higginbottom A., Raman R., Ferraiuolo L., Cooper-Knock J., McDermott C.J., Wharton S.B., Shaw P.J., Ince P.G. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurons. Neuropathol Appl Neurobiol 2014; 40(6): 670–685, http://dx.doi.org/10.1111/nan.12148.
- Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J 2000; 19(17): 4439–4448, http://dx.doi.org/10.1093/emboj/19.17.4439.
- Osborne R.J., Lin X., Welle S., Sobczak K., O’Rourke J.R., Swanson M.S., Thornton C.A. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet 2009; 18(8): 1471–1481, http://dx.doi.org/10.1093/hmg/ddp058.
- Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T., Cline M., Tawil R., Osborne R.J., Wheeler T.M., Swanson M.S., Moxley R.T. 3rd, Thornton C.A. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 2013; 74(6): 862–872, http://dx.doi.org/10.1002/ana.23992.
- Miller D.T., Adam M.P., Aradhya S., Biesecker L.G., Brothman A.R., Carter N.P., Church D.M., Crolla J.A., Eichler E.E., Epstein C.J., Faucett W.A., Feuk L., Friedman J.M., Hamosh A., Jackson L., Kaminsky E.B., Kok K., Krantz I.D., Kuhn R.M., Lee C., Ostell J.M., Rosenberg C., Scherer S.W., Spinner N.B., Stavropoulos D.J., Tepperberg J.H., Thorland E.C., Vermeesch J.R., Waggoner D.J., Watson M.S., Martin C.L., Ledbetter D.H. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010; 86(5): 749–764, http://dx.doi.org/10.1016/j.ajhg.2010.04.006.
- Chong W.W., Lo I.F., Lam S.T., Wang C.C., Luk H.M., Leung T.Y., Choy K.W. Performance of chromosomal microarray for patients with intellectual disabilities/developmental delay, autism, and multiple congenital anomalies in a Chinese cohort. Mol Cytogenet 2014; 7: 34, http://dx.doi.org/10.1186/1755-8166-7-34.
- Emy Dorfman L., Leite J.C., Giugliani R., Riegel M. Microarray-based comparative genomic hybridization analysis in neonates with congenital anomalies: detection of chromosomal imbalances. J Pediatr (Rio J) 2015; 91(1): 59–67, http://dx.doi.org/10.1016/j.jped.2014.05.007.
- Yuen R.K., Merkoulovitch A., MacDonald J.R., Vlasschaert M., Lo K., Grober E., Marshall C.R., Jarvi K.A., Kolomietz E., Scherer S.W. Development of a high-resolution Y-chromosome microarray for improved male infertility diagnosis. Fertil Steril 2014; 101(4): 1079–1085, http://dx.doi.org/10.1016/j.fertnstert.2013.12.027.
- Krepischi A.C., Capelli L.P., Silva A.G., de Araújo É.S., Pearson P.L., Heck B., da Costa C.M., de Camargo B., Rosenberg C. Large germline copy number variations as predisposing factor in childhood neoplasms. Future Oncol 2014; 10(9): 1627–1633, http://dx.doi.org/10.2217/fon.14.41.
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126(4): 663–676, http://dx.doi.org/10.1016/j.cell.2006.07.024.
- Woltjen K., Michael I.P., Mohseni P., Desai R., Mileikovsky M., Hämäläinen R., Cowling R., Wang W., Liu P., Gertsenstein M., Kaji K., Sung H.K., Nagy A. рiggyBack transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009; 458(7239): 766–770, http://dx.doi.org/10.1038/nature07863.
- Lowry W.E., Plath K. The many ways to make an iPS cell. Nat Biotechnol 2008; 26(11): 1246–1248, http://dx.doi.org/10.1038/nbt1108-1246.
- Fathi A., Hatami M., Hajihosseini V., Fattahi F., Kiani S., Baharvand H., Salekdeh G.H. Comprehensive gene expression analysis of human embryonic stem cells during differentiation into neural cells. PLoS One 2011; 6(7): e22856, http://dx.doi.org/10.1371/journal.pone.0022856.
- Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W., Diao Y., Liang J., Zhao H., Lobanenkov V.V., Ecker J.R., Thomson J.A., Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature 2015; 518(7539): 331–336, http://dx.doi.org/10.1038/nature14222.
- Lee J.H., Lee J.B., Shapovalova Z., Fiebig-Comyn A., Mitchell R.R., Laronde S., Szabo E., Benoit Y.D., Bhatia M. Somatic transcriptome priming gates lineage-specific differentiation potential of human-induced pluripotent stem cell states. Nat Commun 2014; 5: 5605, http://dx.doi.org/10.1038/ncomms6605.
- Busskamp V., Lewis N.E., Guye P., Ng A.H., Shipman S.L., Byrne S.M., Sanjana N.E., Murn J., Li Y., Li S., Stadler M., Weiss R., Church G.M. Rapid neurogenesis through transcriptional activation in human stem cells. Mol Syst Biol 2014; 10: 760, http://dx.doi.org/10.15252/msb.20145508.
- Dorn I., Klich K., Arauzo-Bravo M.J., Radstaak M., Santourlidis S., Ghanjati F., Radke T.F., Psathaki O.E., Hargus G., Kramer J., Einhaus M., Kim J.B., Kögler G., Wernet P., Schöler H.R., Schlenke P., Zaehres H. Erythroid differentiation of human induced pluripotent stem cells is independent of donor cell type of origin. Haematologica 2015; 100(1): 32–41, http://dx.doi.org/10.3324/haematol.2014.108068.
- Gabut M., Samavarchi-Tehrani P., Wang X., Slobodeniuc V., O’Hanlon D., Sung H.K., Alvarez M., Talukder S., Pan Q., Mazzoni E.O., Nedelec S., Wichterle H., Woltjen K., Hughes T.R., Zandstra P.W., Nagy A., Wrana J.L., Blencowe B.J. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 2011; 147(1): 132–146, http://dx.doi.org/10.1016/j.cell.2011.08.023.
- Rao S., Zhen S., Roumiantsev S., McDonald L.T., Yuan G.C., Orkin S.H. Differential roles of Sall4 isoforms in embryonic stem cell pluripotency. Mol Cell Biol 2010; 30(22): 5364–5380, http://dx.doi.org/10.1128/MCB.00419-10.
- Han H., Irimia M., Ross P.J., Sung H.K., Alipanahi B., David L., Golipour A., Gabut M., Michael I.P., Nachman E.N., Wang E., Trcka D., Thompson T., O’Hanlon D., Slobodeniuc V., Barbosa-Morais N.L., Burge C.B., Moffat J., Frey B.J., Nagy A., Ellis J., Wrana J.L., Blencowe B.J. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 2013; 498(7453): 241–245, http://dx.doi.org/10.1038/nature12270.
- Wu J.Q., Habegger L., Noisa P., Szekely A., Qiu C., Hutchison S., Raha D., Egholm M., Lin H., Weissman S., Cui W., Gerstein M., Snyder M. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc Natl Acad Sci USA 2010; 107(11): 5254–5259, http://dx.doi.org/10.1073/pnas.0914114107.
- Salomonis N., Schlieve C.R., Pereira L., Wahlquist C., Colas A., Zambon A.C., Vranizan K., Spindler M.J., Pico A.R., Cline M.S., Clark T.A., Williams A., Blume J.E., Samal E., Mercola M., Merrill B.J., Conklin B.R. Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. Proc Natl Acad Sci USA 2010; 107(23): 10514–10519, http://dx.doi.org/10.1073/pnas.0912260107.
- Royo S., Sainz B. Jr., Hernández-Jiménez E., Reyburn H., López-Collazo E., Guerra S. Differential induction of apoptosis, interferon signaling, and phagocytosis in macrophages infected with a panel of attenuated and nonattenuated poxviruses. J Virol 2014; 88(10): 5511–5523, http://dx.doi.org/10.1128/JVI.00468-14.
- Iglesias M.J., Reilly S.J., Emanuelsson O., Sennblad B., Pirmoradian Najafabadi M., Folkersen L., Mälarstig A., Lagergren J., Eriksson P., Hamsten A., Odeberg J. Combined chromatin and expression analysis reveals specific regulatory mechanisms within cytokine genes in the macrophage early immune response. PLoS One 7(2): e32306, http://dx.doi.org/10.1371/journal.pone.0032306.
- Saeed S., Quintin J., Kerstens H.H., Rao N.A., Aghajanirefah A., Matarese F., Cheng S.C., Ratter J., Berentsen K., van der Ent M.A., Sharifi N., Janssen-Megens E.M., Ter Huurne M., Mandoli A., van Schaik T., Ng A., Burden F., Downes K., Frontini M., Kumar V., Giamarellos-Bourboulis E.J., Ouwehand W.H., van der Meer J.W., Joosten L.A., Wijmenga C., Martens J.H., Xavier R.J., Logie C., Netea M.G., Stunnenberg H.G. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014; 345(6204): 1251086, http://dx.doi.org/10.1126/science.1251086.
- Olex A.L., Hiltbold E.M., Leng X., Fetrow J.S. Dynamics of dendritic cell maturation are identified through a novel filtering strategy applied to biological time-course microarray replicates. BMC Immunology 2010; 11: 41, http://dx.doi.org/10.1186/1471-2172-11-41.
- Wang P., Xue Y., Han Y., Lin L., Wu C., Xu S., Jiang Z., Xu J., Liu Q., Cao X. The STAT3-binding long noncoding RNA Inc-DC controls human dendritic cell differentiation. Science 2014; 344(6181): 310–313, http://dx.doi.org/10.1126/science.1251456.
- Zak D.E., Andersen-Nissen E., Peterson E.R., Sato A., Hamilton M.K., Borgerding J., Krishnamurty A.T., Chang J.T., Adams D.J., Hensley T.R., Salter A.I., Morgan C.A., Duerr A.C., De Rosa S.C., Aderem A., McElrath M.J. Merck Ad5/HIV induces broad innate immune activation that predicts CD8+ T-cell responses but is attenuated by preexisting Ad5 immunity. Proc Natl Acad Sci USA 2012; 109(50): E3503–E3512, http://dx.doi.org/10.1073/pnas.1208972109.
- Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C., Wang E., Douek D.C., Price D.A., June C.H., Marincola F.M., Roederer M., Restifo N.P. A human memory T cell subset with stem cell-like properties. Nat Med 2011; 17(10): 1290–1298, http://dx.doi.org/10.1038/nm.2446.
- Kolokoltsova O.A., Yun N.E., Paessler S. Reactive astrogliosis in response to hemorrhagic fever virus: microarray profile of Junin virus-infected human astrocytes. Virol J 2014; 11: 126, http://dx.doi.org/10.1186/1743-422X-11-126.
- D’Aiuto L., Prasad K.M., Upton C.H., Viggiano L., Milosevic J., Raimondi G., McClain L., Chowdari K., Tischfield J., Sheldon M., Moore J.C., Yolken R.H., Kinchington P.R., Nimgaonkar V.L. Persistent Infection by HSV-1 is associated with changes in functional architecture of iPSC-derived neurons and brain activation patterns underlying working memory performance. Schizophr Bull 2015; 41(1): 123–132, http://dx.doi.org/10.1093/schbul/sbu032.
- Thomas E., Gonzalez V.D., Li Q., Modi A.A., Chen W., Noureddin M., Rotman Y., Liang T.J. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 2012; 142(4): 978–988, http://dx.doi.org/10.1053/j.gastro.2011.12.055.
- Zamanian J.L., Xu L., Foo L.C., Nouri N., Zhou L., Giffard R.G., Barres B.A. Genomic analysis of reactive astrogliosis. J Neuroscience 2012; 32(18): 6391–6410, http://dx.doi.org/10.1523/JNEUROSCI.6221-11.2012.
- Shulzhenko N., Morgun A., Hsiao W., Battle M., Yao M., Gavrilova O., Orandle M., Mayer L., Macpherson A.J., McCoy K.D., Fraser-Liggett C., Matzinger P. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med 2011; 17(12): 1585–1594, http://dx.doi.org/10.1038/nm.2505.
- Rodrigues R., Grosso A.R., Moita L. Genome-wide analysis of alternative splicing during dendritic cell response to a bacterial challenge. PLoS One 2013; 8(4): e61975, http://dx.doi.org/10.1371/journal.pone.0061975.
- Ip J.Y., Tong A., Pan Q., Topp J.D., Blencowe B.J., Lynch K.W. Global analysis of alternative splicing during T-cell activation. RNA 2007; 13(4): 563–572, http://dx.doi.org/10.1261/rna.457207.








