Molecular Markers of EBV- and HHV6-Associated Mononucleosis
Epstein–Barr virus (EBV) and human herpesvirus type 6 (HHV6) are causative agents of infectious mononucleosis and can lead to the development of lymphoproliferative diseases. Means of radical therapy for this disease are yet to be found. Key transcripts involved in the pathogenesis can be used as molecular markers and also as potential therapeutic targets.
The aim of the study was to identify molecular markers associated with infection caused by EBV and HHV6; specifically, we looked into the markers localized in blood leukocytes of patients with infectious mononucleosis.
Materials and Methods. We studied the transcriptome of peripheral blood leukocytes in children and adolescents with infectious mononucleosis caused by Epstein–Barr virus (EBV-IM) and human herpesvirus type 6 (HHV6-IM), as well as healthy subjects matched by gender and age. Using our original DNA biochips, we determined the expression of 403 genes (total representation level of all mRNA of one gene) and 712 transcripts (individual spliced mRNA of one gene) essential for the proliferation and apoptosis of immunocompetent cells. Data analysis was performed using a combination of machine learning and traditional statistics. The genes and transcripts which are highly important for paired classification and have the statistically significant differences in the expression between patients and healthy subjects were selected to serve molecular markers of the infection.
Results. Unique groups of candidate markers for EBV-IM and HHV6-IM were identified. EBV-IM was characterized by a decreased expression of the AR transcript 5 and ASCC1 transcript 4 and also of the CAD gene and FADD mRNA; an increased expression of the HLA-DPA1 transcript 2 and RIPK1 transcript 4 were found. In patients with HHV6-IM, an increase in the expression of AVEN mRNA, CHUK transcript 2, CIRBP transcript 2, and TRAF3 transcript 2, as well as a decrease in the expression of IRAK4 transcript 10 was observed. In the post-infection period, the expression levels of most of the markers returned to normal.
Conclusion. The sets of identified markers are uniquely characteristic of the two infections (EBV-IM and HHV6-IM) and can be used as targets for new therapies.
Introduction
Epstein–Barr virus (EBV, Human gammaherpesvirus 4) and human herpesvirus type 6 (HHV6, Human betaherpesvirus 6) are members of the Herpesviridae family that cause infectious mononucleosis (IM). Both viruses have tropism for various immunocompetent cells, can change the number of these cells in the blood [1], and contribute to the development of lymphoproliferative diseases [2–4]. To date, there are no effective means of targeted therapy for the disease.
At the molecular level, these viruses modulate the expression of genes involved in the activation of immunocompetent cells, their proliferation, differentiation, and apoptosis. A pronounced change in the collective expression of the genes controlling these processes can serve as the unique molecular passport of EBV-IM and HHV6-IM, and the expression of individual genes — as markers of the infection. Such markers can be used to diagnose and monitor the disease, and also represent potential therapeutic targets [5–8].
Using the DNA biochip technology (DNA microarray), it is possible to evaluate the expression of an individual gene (represented by its total mRNA level), and also identify and quantify its transcripts (spliced mRNAs). Considering the optimal ratio of research cost, productivity and accuracy, DNA biochips often become the first choice technology for detecting genetic markers of a disease [9]. To analyze the results obtained with DNA biochips or with high-performance sequencing methods, a complex mathematical apparatus that combines applied statistics and machine learning algorithms is required [10].
The aim of this study was to identify molecular markers of EBV-IM and HHV6-IM expressed in blood leukocytes of patients with infectious mononucleosis.
Materials and Methods
Design and synthesis of the DNA biochip. The expression of mRNA of genes of interest was assayed by using DNA biochips designed in this laboratory. We selected the discriminating probes — the functional basis of the biochip — with the help of the “Splice variants microarray design pipeline” algorithm [11]. A total of 1115 probes were selected; of those, 403 allowed us to measure the gene expression, and the rest 712 probes were able to detect the presence of individual transcripts. A complete list of genes and their transcripts is presented in Appendix 1, https://drive.google.com/file/d/1xKS_MvENloPYKwuzeC1LaqFTH3i130So/view. In addition, 70 negative control probes, selected on the basis of the genome of Rhizobium rubi bacterium, were incorporated in the DNA biochip. Biochip probes were synthesized in situ using the B3 Synthesizer equipped with the relevant reagent kit in accordance with the manufacturer’s recommendations (CustomArray Inc., USA).
Selection of study groups. The study included children and adolescents 7–18 years old diagnosed with acute infectious mononucleosis, as well as practically healthy volunteers matched by gender and age. In the blood and serum of the participants, the presence of specific antibodies to EBV, HHV6, and cytomegalovirus (CMV), as well as the presence of DNA of these pathogens were determined. For this purpose, we used commercial kits for immunoassay diagnostics: VectoEBV-VCA-IgM, VectoEBV-VCA-IgG, VectoHHV-6-IgG, VectoCMV-IgM, VectoCMV-IgG (Vector-Best, Russia) and a commercial real-time PCR kit “AmpliSense EBV/CMV/HHV6-screen-FL” (Central Research Institute of Epidemiology, Russia).
According to the test results, the following groups were identified: NORM — practically healthy subjects without clinical or laboratory signs of infection (n=17, average age — 11 years), EBVinf — patients with EBV-associated IM (n=6, average age — 12 years), and HHV6inf — patients with HHV6-associated IM (n=7, mean age — 11 years). Clinically healthy controls with laboratory signs of infection, patients with CMV-mediated IM or mixed infection were excluded from the study.
Blood sampling. Peripheral blood samples were used in the study. Blood samples from the infected patients were taken before the start of the treatment, as well as after the recovery and disappearance of clinical and laboratory signs of the disease (EBVrec and HHV6rec groups). The recovery period samples were taken (on average) 2 months upon the resolution of the disease.
The study was conducted in accordance with the Helsinki Declaration (2013) and approved by the Ethics Committee of the Blokhina Scientific Research Institute of Epidemiology and Microbiology of Nizhny Novgorod. The samples were taken after informed consent was obtained from children’s parents or guardians.
Preparation and hybridization of mRNA. To isolate leukocyte from the blood, we used the Hemolysis solution (Central Research Institute of Epidemiology, Russia). Using the MAGNO-sorb kit (Central Research Institute of Epidemiology, Russia), a total RNA pool was isolated from the obtained leukocytes, which was further purified and concentrated with a mixture of phenol and chloroform in a 1:1 ratio. Total RNA (2 µg) then underwent reverse transcription and addition of the second strand by using the Mint cDNA synthesis kit (Evrogen, Russia), while the 3’-oligo-T-primer was replaced by the T7 containing promoter-oligo-T-primer (DNA Synthesis, Russia). The completion of the second strand and the amplification of cDNA was performed with 15 cycles of PCR: 95°C — 25 s; 60°C — 25 s; 72°C — 6 min. The resulting double-stranded cDNA (2 μg) was transcribed using T7 RNA polymerase (Thermo Scientific, England), under the addition of the biotin label in the form of biotinylated UTP nucleotides (DNA Synthesis, Russia). The transcription procedure was repeated three times.
The labeled RNA samples resulted from the synthesis were hybridized separately. The RNA hybridization procedure on the biochip and the amperometric readings were performed using ElectraSense equipment and reagents (CustomArray Inc., USA) in accordance with the manufacturer’s recommendations. The resulting signal was considered as the relative expression of the gene or transcript.
Data processing. Data processing and calculations were performed using the R programming language (Version 3.5.0; R Foundation for Statistical Computing, Austria) and RStudio environment (version 1.1.453; RStudio, USA). The data were normalized using quantile normalization based on the negative control [12]. Then, a matrix of results was created where the rows corresponded to the factors — levels of gene and transcript expression, and the columns — to RNA samples from one of the examined groups (NORM, EBVinf, HHV6inf, EBVrec, and HHV6rec).
Further, the pairwise discriminant analysis was performed for all five groups. In this step, a model of the relation between the given RNA sample and one of the two compared groups was created. Here, we used the algorithm of gradient boosting over the decision trees. The model parameters were selected and its performance was evaluated using the cross-validation method. The resulting classifier was considered effective when the median value of the accuracy and the median of the AUC — area under the error curve (area under the receiver operating characteristic (ROC) curve) was not less than 0.75. In the case the classifier proved effective, we improved the quality of the classification by repeating the modeling procedure and reducing the number of factors. We selected 20 genes or transcripts of the highest importance (feature importance) for each model obtained from the cross-validation [10]. Further, the selected indicators were used to repeat the modeling procedure, which was implemented similarly to the first one.
At the next stage, the expression level of each gene or transcript was compared between the groups NORM vs EBVinf, NORM vs HHV6inf, EBVinf vs HHV6inf, NORM vs EBVrec, and NORM vs HHV6rec. A t-test was applied after the adjustment for the expected false discovery rate (FDR). At q<0.05, differences in the expression of genes and transcripts were considered statistically significant.
Changes in the level of expression were also calculated. In each group of patients, the average value of gene/transcript expression was determined; then, the difference in the expression was calculated for each pair of groups (%):
(group 2 average · 100 / group 1 average) – 100.
Based on the importance of the examined genes and transcripts for re-classification and also on the changes in their expression level and statistical significance of these changes, we decided on whether the given factor could be classified as a molecular marker of EBV-IM or HHV6-IM. For the NORM–EBVinf pair, we selected genes and transcripts that met the following requirements: the expression level of the marker in the EBVinf group was significantly different from that in the NORM group; this difference in the expression and/or the importance of the marker for the NORM–EBVinf classification exceeded the 75th percentile (the threshold value was established empirically) of the respective parameter for all factors; the marker was part of the classification factors for HHV6inf–EBVinf pair; the marker was not part of the classification factors for the NORM–HHV6inf pair. The genes or transcripts selected through this procedure were considered markers for EBV infection. The HHV6 infection markers were selected similarly from the NORM–HHV6inf classification factors.
Results
At the initial stage, models for the classification of mRNA samples in the NORM–EBVinf, NORM–HHV6inf, and HHV6inf–EBVinf paired groups were developed. In parallel, we reduced the number of factors and thus improved the quality of these models at the next stage when the NORM–EBVinf and NORM–HHV6inf groups underwent the re-classification. For the comparison groups of HHV6inf–EBVinf, the reduction in the number of factors and the re-modeling had little effect on the quality of classification (Table 1). On average, the number of factors used for the second classification was less than 8% of the initial set. The complete list of genes and transcripts considered for the re-modeling of the RNA sample relation to the NORM–EBVinf, NORM–HHV6inf, and HHV6inf–EBVinf groups, is presented in the appended materials (see Appendices 2–4, https://drive.google.com/file/d/1xKS_MvENloPYKwuzeC1LaqFTH3i130So/view). For the comparison groups, EBVinf–EBVrec, HHV6inf–HHV6rec, NORM–EBVrec, and NORM-HHV6rec, we were unable to create satisfactory models of the RNA sample relation to any of the groups (see Table 1).
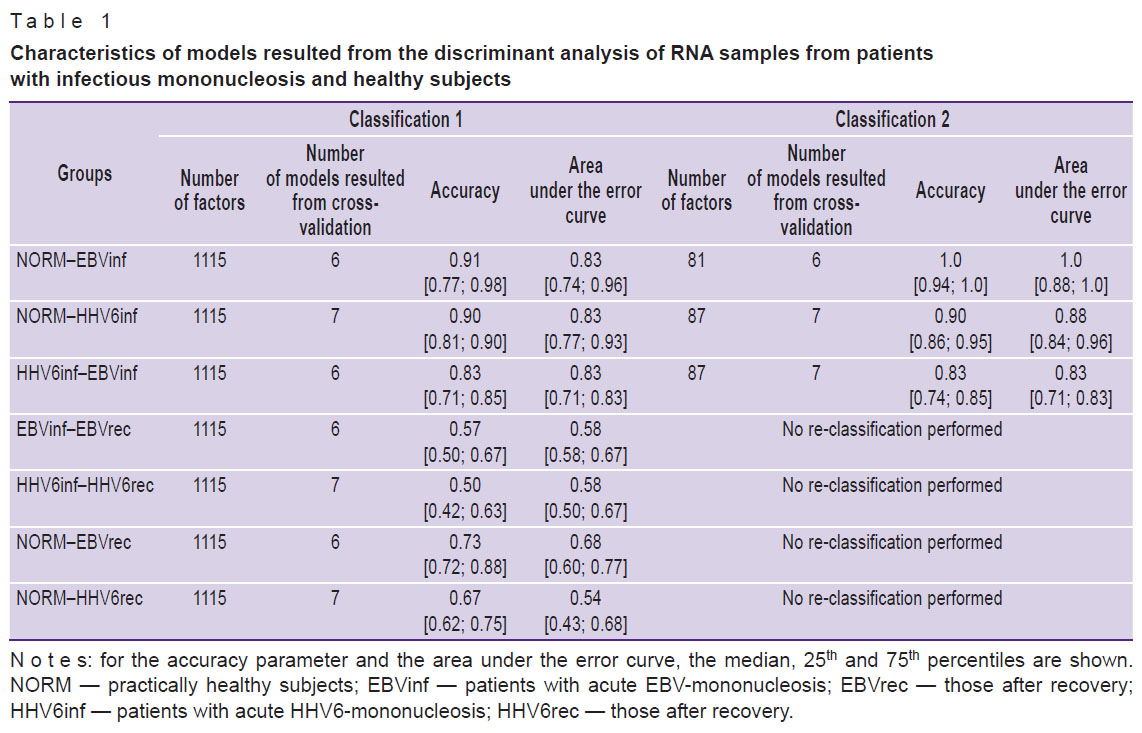
|
Table 1. Characteristics of models resulted from the discriminant analysis of RNA samples from patients with infectious mononucleosis and healthy subjects |
Further, according to the algorithm, unique sets of molecular markers allowing us to distinguish between EBV-IM and HHV6-IM were selected (see the Figure). For each of the identified markers, changes in the level of its expression were compared between normal subjects and patients with IM in the acute phase and the recovery period (Table 2). In patients with acute EBV-IM, there was a decrease in the expression of the AR transcript 5 and ASCC1 transcript 4, FADD mRNA, and also the CAD gene as compared to the norm. On the contrary, the expression of HLA-DPA1 transcript 2 and RIPK1 transcript 4 increased. The process of patient recovery was characterized by the return of all indicators to the normal levels, except for the expression of the CAD gene and the RIPK1 transcript 4, which remained abnormal. In patients with HHV6-IM, regardless of the phase of the disease, the expression of EBV-IM markers corresponded to that in healthy subjects. The exception was FADD mRNA: its expression in patients with HHV6-IM increased.
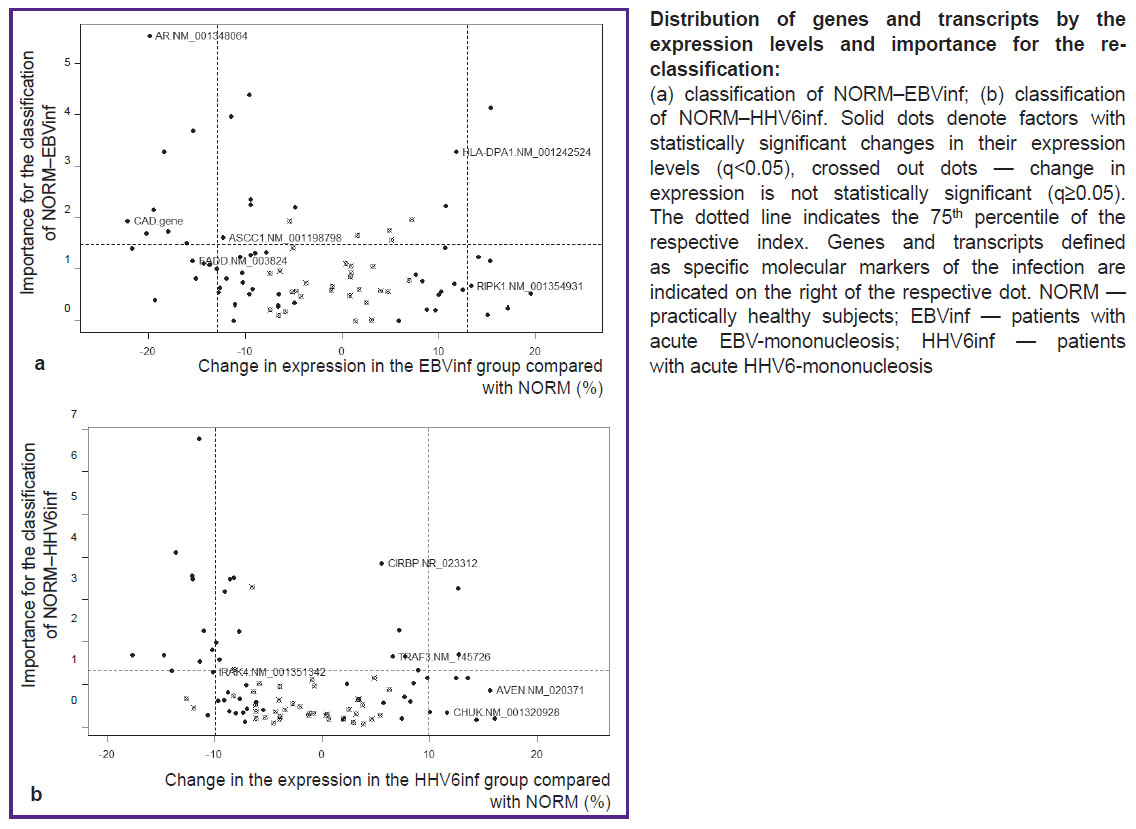 |
Distribution of genes and transcripts by the expression levels and importance for the re-classification: (a) classification of NORM–EBVinf; (b) classification of NORM–HHV6inf. Solid dots denote factors with statistically significant changes in their expression levels (q<0.05), crossed out dots — change in expression is not statistically significant (q≥0.05). The dotted line indicates the 75th percentile of the respective index. Genes and transcripts defined as specific molecular markers of the infection are indicated on the right of the respective dot. NORM — practically healthy subjects; EBVinf — patients with acute EBV-mononucleosis; HHV6inf — patients with acute HHV6-mononucleosis |
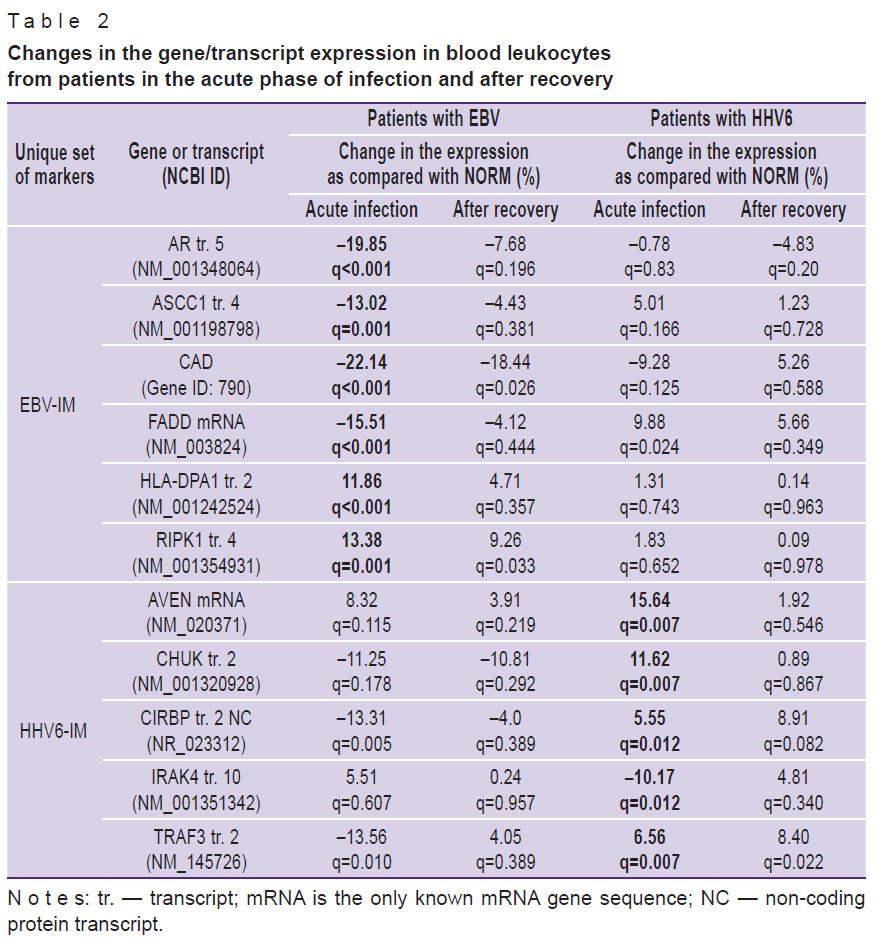
|
Table 2. Changes in the gene/transcript expression in blood leukocytes from patients in the acute phase of infection and after recovery |
In the acute period of HHV6-IM, an increase in the expression of the AVEN mRNA, CHUK transcript 2, CIRBP transcript 2, and TRAF3 transcript 2, as well as a decrease in the expression of the IRAK4 transcript 10 was detected. After recovery, normalization of all parameters was observed except for the expression level of the TRAF3 transcript 2, which remained elevated. In patients with EBV-IM, the expression of the above markers did not differ from the norm, with the exception of the CIRBP transcript 2 and IRAK4 transcript 10: their presence decreased (CIRBP transcript 2) and increased (IRAK4 transcript 10) during the acute phase of infection.
Discussion
The algorithm we propose to process the data on the hybridization of RNA samples to DNA biochip, allows us to identify a unique set of molecular markers of EBV-IM and HHV6-IM. This algorithm is based on a combination of machine learning and traditional statistics. Such a combined approach made it possible to solve several tasks: a) demonstrate the feasibility of grouping the patients by the expression of certain genes and individual transcripts (the task of classification), b) reduce the number of analyzed factors by removing the elements insignificant for the analysis (selection of parameters important for classification), c) identify statistically significant changes in the expression of each of the many genes in patients with EBV-IM and HHV6-IM (the task of statistics), and d) select a set of genes and transcripts, which is unique for each of the diseases (logical exception).
In this study, we created classifiers that allowed us to discern between practically healthy individuals, patients with EBV-IM and those with HHV6-IM; these classifiers are based on the expression of certain genes and transcripts. This classification is indicative of differences in the pathogenetic mechanisms of the two virus-associated infections. On the other hand, it was impossible to create the models of the difference in the gene or transcript expression between post-IM patients, healthy subjects, and patients with acute IM. The result suggests that the expression of the selected genes and transcripts does not return to normal for at least two months after clinical recovery, and thus maintains the “molecular footprint” of the infection. This conclusion is confirmed by the fact that some of the EBV-IM and HHV6-IM markers are expressed at abnormal levels even after recovery (see Table 2). Therefore, EBV and HHV6 may have a prolonged effect on the patient’s immune system by regulating the expression of genes and transcripts involved in apoptosis, proliferation, and other vital processes in immunocompetent cells.
Using the proposed algorithm, we have identified unique sets of molecular markers for EBV- and HHV6-associated mononucleosis. The overwhelming majority of the candidate markers are spliced mRNAs of genes belonging to different functional groups. Some transcripts are the only known or “basic” form of mRNA of gene (FADD mRNA, AVEN mRNA). Usually, the function of proteins encoded by such transcripts is described in the literature as a property of the gene itself. Other transcripts encode structural and/or functional forms of a protein, different from the “basic” form, but exhibiting the similar properties (AR transcript 5, CHUK transcript 2, and IRAK4 transcript 10). The third group includes spliced mRNAs of a gene, which differ from the “basic” form by the nucleotide sequences in the 5’-NTR (HLA-DPA1 transcript 2 and RIPK1 transcript 4). The protein product of such transcripts is similar to the product of the “basic” spliced form of mRNA. At the same time, the stability of the transcripts themselves and the productivity of their translation may differ due to the differences in the nucleotide composition in the regulatory site of the mRNA. Of particular interest are transcripts whose translation products have a function that differs from that of the “basic” variant of mRNA or is directly opposite to it. Among the markers identified in this study is the ASCC1 transcript 4, which encodes a protein that, unlike the “basic” form, is not capable of inhibiting the expression of NF-kB and NF-kB-targeted genes [13]. Another example is the transcript 2 of the NF-kB kinase inhibitor TRAF3, whose product induces the expression of NF-kB in activated T-cells [14].
In addition to protein-coding transcripts, we identified the non-coding transcript 2 of CIRBP — a cell cycle regulator and an inducer of apoptosis. The functional significance of non-protein-translating transcripts is well explained in terms of the concept of unproductive splicing, i.e., an alternative splicing that leads to the formation of non-coding transcripts of the target gene and their subsequent degradation. The change in the ratio of coding to non-coding transcripts determines the expression level of the protein product of the target gene [15]. The described mechanism of the gene expression control through alternative splicing can be extended to protein-coding variants of mRNA: an increase in the proportion of one transcript leads to a decrease in the proportion of other transcripts of the same gene and vice versa.
Thus, the use of transcripts as markers of an infectious disease is not only justified, but also promising in terms of expanding the diagnostic and prognostic capabilities of biomedicine. Different transcripts of the same gene may convey different clinical information. Thus, according to our data, a decrease in the expression of the ASCC1 transcript 4 is specific only for EBV-IM, while a change in the expression of the ASCC1 transcript 1 is detected both in EBV-IM and in HHV6-IM (see Appendices 2, 3, https://drive.google.com/file/d/1xKS_MvENloPYKwuzeC1LaqFTH3i130So/view). Most of the currently used diagnostic test systems determine the gene expression or protein content, but not the ratio between individual transcripts.
Our results imply that the proliferation and apoptosis in the blood leukocytes of patients with EBV-IM and HHV6-IM are regulated in different ways as compared to healthy controls. In EBV infection, we observed a decrease in the expression of proliferation promoters, such as transcript 5 of the steroid hormone receptor AR, the ASCC1 transcript 4, and the CAD gene. On the other hand, in EBV-IM there was an increase in the expression of the RIPK1 transcript 4 and a decrease in the adapter FADD mRNA expression; the FADD molecule plays a key role in the cytotoxic immune response and apoptosis. RIPK1 kinase is a multifunctional protein that is part of signaling pathways of necroptosis, inflammation, and NF-kB activation. It is notable that the development of necroptosis requires the formation of a large signal complex, which includes equimolar amounts of RIPK1, FADD, caspase-8, and other proteins [16]. Therefore, the increased expression of RIPK1 transcript 4 in EBV infection against the background of decreased FADD expression can inhibit apoptosis and promote cell survival and proliferation. In addition, we found an increase in the expression of the HLA-DPA1 transcript 2, which not only plays an important role in the antigen presentation but also serves as a co-receptor that facilitates EBV entry into B-lymphocytes [17].
The functional role of HHV6-IM markers is largely associated with activation of transcription and suppression of apoptosis. Thus, an increase in the expression of NF-kB activators of the TRAF3 transcript 2 and the CHUK transcript 2 was observed against the background of an increased expression of mRNA of AVEN — a mitochondrial apoptosis inhibitor. The expression of the non-coding transcript 2 of the proapoptotic CIRBP factor increased, which also indicated a decrease in the apoptotic activity in the cells. In addition, we note a decrease in the expression of the IRAK4 transcript 10, which is an important mediator of the innate immune response.
The identified unique sets of molecular markers of EBV- and HHV6-associated infectious mononucleosis can be used as therapeutic targets in the development of targeted biotherapy. It is important to note that the proposed algorithm can be used to search for unique markers of other infectious diseases.
Conclusion
In blood leukocytes of children and adolescents with EBV- and HHV6-associated mononucleosis, a change in the expression of several genes and transcripts regulating the activation, proliferation, and apoptosis of immunocompetent cells was detected. Unlike healthy subjects, leukocytes from patients with EBV infection had a decreased expression of the AR transcript 5 and ASCC1 transcript 4, the CAD gene and FADD mRNA against the background of an increased expression of the HLA-DPA1 transcript 2 and RIPK1 transcript 4. In patients with HHV6-IM, an increase in the expression of AVEN mRNA, CHUK transcript 2, CIRBP transcript 2, and TRAF3 transcript 2, as well as a decrease in the expression of IRAK4 transcript 10 was detected. The identified markers are known to play a role in the pathogenesis of EBV-IM and HHV6-IM and reflect specific features of their molecular mechanisms, as well as the immune response to the infection. The sets of identified markers are unique for the two infections under study.
Research funding. The study was supported by grant No.AAAA-A16-116040810135-4.
Conflict of interest. The authors declare no conflict of interest.
References
- Filatova E.N., Solntsev L.A., Presnyakova N.B., Kulova E.A., Utkin O.V. Determination of some immunological features of hhv-6-mediated infectious mononucleosis in children by the method of discriminatory analysis. Infektsiya i immunitet 2018; 8(2): 223–229, https://doi.org/10.15789/2220-7619-2018-2-223-229.
- Dojcinov S., Fend F., Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts. Pathogens 2018; 7(1): 28, https://doi.org/10.3390/pathogens7010028.
- Nakayama-Ichiyama S., Yokote T., Oka S., Iwaki K., Kobayashi K., Hirata Y., Hiraoka N., Takayama A., Akioka T., Miyoshi T., Takubo T., Tsuji M., Hanafusa T. Diffuse large B-cell lymphoma, not otherwise specified, associated with coinfection of human herpesvirus 6 and 8. J Clin Oncol 2011; 29(21): e636–e637, https://doi.org/10.1200/jco.2011.35.1254.
- Razzaque A. Oncogenic potential of human herpesvirus-6 DNA. Oncogene 1990; 5(9): 1365–1370.
- Li B., Zeng Q. Personalized identification of differentially expressed pathways in pediatric sepsis. Mol Med Rep 2017; 16(4): 5085–5090, https://doi.org/10.3892/mmr.2017.7217.
- Omar M., Klawonn F., Brand S., Stiesch M., Krettek C., Eberhard J. Transcriptome-wide high-density microarray analysis reveals differential gene transcription in periprosthetic tissue from hips with chronic periprosthetic joint infection vs aseptic loosening. J Arthroplasty 2017; 32(1): 2342–2340, https://doi.org/10.1016/j.arth.2016.06.036.
- Sano D., Tazawa M., Inaba M., Kadoya S., Watanabe R., Miura T., Kitajima M., Okabe S. Selection of cellular genetic markers for the detection of infectious poliovirus. J Appl Microbiol 2018; 124(4): 1001–107, https://doi.org/10.1111/jam.13621.
- Scicluna B.P., van Vught L.A., Zwinderman A.H., Wiewel M.A., Davenport E.E., Burnham K.L., Nürnberg P., Schultz M.J., Horn J., Cremer O.L., Bonten M.J., Hinds C.J., Wong H.R., Knight J.C., van der Poll T.; MARS consortium. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med 2017; 5(10): 816–826, https://doi.org/10.1016/s2213-2600(17)30294-1.
- Knyazev D.I., Starikova V.D., Utkin О.V., Solntsev L.А., Sakharnov N.А., Efimov E.I. Splicing-sensitive DNA-microarrays: peculiarities and applicationin biomedical research (review). Sovremennye tehnologii v medicine 2015; 7(4): 162–173, https://doi.org/10.17691/stm2015.7.4.23.
- Pirooznia M., Yang J.Y., Yang M.Q., Deng Y. A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics 2008; 9(Suppl 1): S13, https://doi.org/10.1186/1471-2164-9-s1-s13.
- Solntsev L.A., Starikova V.D., Sakharnov N.A., Knyazev D.I., Utkin O.V. Strategy of probe selection for studying mRNAs that participate in receptor-mediated apoptosis signaling. Mol Biol 2015; 49(3): 457–465, https://doi.org/10.1134/s0026893315030164.
- Wu Z., Aryee M.J. Subset quantile normalization using negative control features. J Comput Biol 2010; 17(10): 1385–1395, https://doi.org/10.1089/cmb.2010.0049.
- Torices S., Alvarez-Rodríguez L., Grande L., Varela I., Muñoz P., Pascual D., Balsa A., López-Hoyos M., Martinez-Taboada V., Fernández-Luna J.L. A truncated variant of ASCC1, a novel inhibitor of NF-B, is associated with disease severity in patients with rheumatoid arthritis. J Immunol 2015; 195(11): 5415–5420, https://doi.org/10.4049/jimmunol.1501532.
- Michel M., Wilhelmi I., Schultz A.-S., Preussner M., Heyd F. Activation-induced tumor necrosis factor receptor-associated factor 3 (Traf3) alternative splicing controls the noncanonical nuclear factor κB pathway and chemokine expression in human T cells. J Biol Chem 2014; 289(19): 13651–13660, https://doi.org/10.1074/jbc.m113.526269.
- Filatova E.N., Utkin O.V. The role of noncoding MRNA isoforms in the regulation of gene expression. Russ J Genet 2018; 54(8): 879–887, https://doi.org/10.1134/s1022795418080057.
- Feoktistova M., Leverkus M. Programmed necrosis and necroptosis signalling. FEBS J 2014; 282(1): 19–31, https://doi.org/10.1111/febs.13120.
- Haan K.M., Kwok W.W., Longnecker R., Speck P. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J Virol 2000; 74(5): 2451–2454, https://doi.org/10.1128/jvi.74.5.2451-2454.2000.









